
Dr. Ulises D. Lima Rodríguez,1 Dr. Antonio Raunel Hernández Rodríguez,2 Dra. Lina Marta Pérez Espinosa3 y Dra. Marianela Alberro Fernández3
Lima Rodríguez UD, Hernández Rodríguez AR, Pérez Espinosa LM, Alberro Fernández M. Osteogénesis imperfecta tipo II. Reporte de 1 caso. Rev Cubana Ortop Traumatol 1999;13(1-2):115-8.
Se presenta el caso de un nacido muerto con deformidades en miembros inferiores de una gestante de 32 años. Se realizó estudio y descripción clínica del hábito externo y se detectaron las anormalidades siguientes: Cráneo: fontanelas amplias; Cara: ojos con órbitas poco profundas, escleróticas azules, puente nasal deprimido y deformidades en miembros inferiores. En el estudio radiográfico se observaron múltiples fracturas en las costillas (imagen arrosariada) y fémur (imagen de acordeón). El estudio clínico de los padres fue normal. Se planteó el diagnóstico de una osteogénesis imperfecta tipo II, que probablemente tenga como causa una mutación fresca en el material hereditario de uno de los padres. La pareja recibió atención multidisciplinaria que incluyó el asesoramiento genético y han decidido no tener más hijos por el momento.
Descriptores DeCS: OSTEOGENESIS IMPERFECTA/genética; ANOMALIAS MULTIPLES; OSTEOGENESIS IMPERFECTA/diagnóstico; MUERTE FETAL.
La osteogénesis imperfecta (OI) es una enfermedad que presenta heterogeneidad genética, por tanto su aspecto clínico depende del tipo específico de que se trate. El cuadro clínico puede variar desde un lactante con un trastorno congénito letal hasta el de un adulto que presente una ligera tendencia a las fracturas óseas. Su diagnóstico es principalmente clínico. Sillence la clasifica en tipos que van desde el I al IV.1,2
La aparición de OI tipo II o letal en hijos de padres sanos ya estaba bien descrita por Remigo PA, en 19703 y McKusick VA, en 1972.4 El síndrome de OI tipo II está en el grupo de los trastornos del tejido conectivo y se caracteriza por: múltiples fracturas, huesos largos acortados y anchos y escleróticas azules. Otras anormalidades descritas son déficit prenatal en el crecimiento de los miembros, cráneo con pobre mineralización, fontanelas amplias, múltiples huesos wormianos, órbitas poco profundas, nariz pequeña y puente nasal deprimido. En los estudios radiográficos se ven las costillas finas con múltiples fracturas, callos óseos, especialmente en miembros inferiores.5 Aunque los términos "letal perinatal" y "congénita" se han usado para este tipo de OI, con un estudio radiográfico cuidadoso pueden distinguirse 3 subtipos:
Se realizó el estudio clínico y radiográfico del producto de la concepción de una embarazada de 32 años de edad con una historia obstétrica de 2 embarazos: el primero con un parto eutócico y un niño normal; el segundo, un parto distócico a las 40 semanas que culminó en una operación cesárea. Se extrajo un nacido muerto del sexo masculino que pesó 2 900 g y que presentaba en su hábito externo como signo fundamental, los miembros inferiores deformados y acortados. Se realizó examen radiográfico de cuerpo entero en vista anteroposterior y de miembros inferiores.
El resultado del estudio del hábito externo mostró las siguientes anormalidades: Cráneo: fon-tanelas amplias. Cara: órbitas poco profundas, escleróticas azules, nariz pequeña con puente nasal deprimido. Miembros inferiores: deformados y acortados (figs. 1 y 2). En el estudio radiográfico se pudo observar el cráneo con pobre mineralización, costillas finas con múltiples fracturas con aspecto arrosariado debido a la gran cantidad de callos óseos; miembros con los huesos largos engrosados y también con múltiples fracturas, sobre todo en los fémures, que le daban un aspecto de acordeón.
Fig. 1. Nacido muerto con osteogénesis imperfecta tipo II.
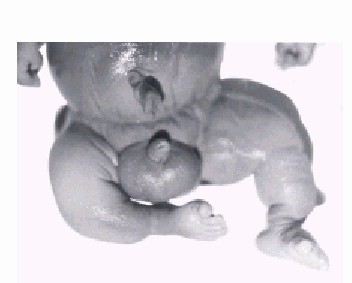
Fig. 2. Miembros inferiores deformados y acortados por múltiples fracturas.
El estudio clínico y radiográfico coincidió con el diagnóstico de OI tipo II. La pareja se atendió por un equipo multidisciplinario. Fueron examinados clínicamente con el objetivo de descubrir signos como baja estatura o piel de textura muy suave13 que nos orientaran a que uno de ellos fuera un mosaico para la mutación génica; el examen físico de los dos fue normal; además, recibieron asesoramiento genético y por ahora decidieron evitar un nuevo embarazo.
La OI tipo II presenta una frecuencia relativamente baja en la población.9 Su causa más frecuente es una mutación fresca en el material hereditario del gameto de uno de los progenitores por lo que tiene un riesgo de recurrencia relativamente bajo, alrededor de 6 %.5 Si fuera causada por un gen recesivo autosómico presente en doble dosis, porque ambos padres fueran heterocigotos, entonces el riesgo de recurrencia aumentaría de 10 a 25 % para los siguientes embarazos.6 Pero si es debido a que un progenitor presenta un mosaicismo para el gen mutado dominante autosómico, donde unas líneas celulares lo tienen y otras no, éste pudiera presentar signos clínicos ligeros de esta enfermedad como baja estatura y textura de la piel suave,14 entonces en este caso el riesgo de recurrencia pudiera elevarse hasta 50 %.
]]> Uno de los requisitos fundamentales para el asesoramiento genético es el diagnóstico certero, por tanto el diagnóstico de la OI tipo II en este caso nos permitió el manejo de esta pareja, pronosticar su bajo riesgo de recurrencia y su posterior seguimiento si su conducta reproductiva varía y desearan un nuevo embarazo.The case of a still birth with deformities in the lower limbs, whose mother was 32 years old, is presented. The following anomalies were detected by the study and clinical description of the external habit: Skull: wide fontanelle; Face: eyes with little deep orbits, blue sclerotics, depressed bridge of the nose and deformities of the lower limbs. Multiple fractures of the ribs (rosary-like image) and femur (accordeon image) were observed in the radiographic study. The clinical study of the parents was normal. It was made the diagnosis of a type II osteogenesis imperfecta probably caused by a fresh mutation in the hereditary material of one of the parents. The couple received multidisciplinary attention that included genetic counselling and they have decided not to have children for the time being.
Subject headings: OSTEOGENESIS IMPERFECTA/genetics; ABNORMALITIES, MULTIPLE; OSTEOGENESIS IMPERFECTA/diagnosis; FETAL DEATH.
Le cas dun foetus mort, avec des difformités des membres inférieurs, dune femme enceinte âgée de 32 ans, est présenté. On a effectué une étude et une description clinique du habitus externe, et les anormalités suivantes ont été détectées: Crâne: larges fontanelles; Visage: yeux à orbites peu profondes, sclérotiques de couleur bleue, pont nasal enfoncé et difformités des membres inférieurs. Dans létude radiographique, on a constaté de multiples fractures sur les côtes (image sous forme de chapelet) et le fémur (image sous forme d-accordéon). Létude clinique des parents a été normale. Le diagnostic dune ostéogenèse imparfaite type II, étant probablement à lorigine dune nouvelle mutation dans le matériel héréditaire dun des parents, a été exposé. Le couple a reçu des soins pluridisciplinaires, y compris lassistance génetique, et ils ont décidé de ne plus avoir des enfants pour le moment.
Mots clés: OSTEOGENESE IMPARFAITE/génétique; ANOMALIES MULTIPLEES; OSTEOGENESE IMPARFAITE/diagnostic; MORT FETALE.
1. Sillence DO, Senn A, Canks DM. Clinical heterogeneity in osteogenesis imperfecta. J Med Genet 1979;16:101-16.
2. Sillence DO. Osteogénesis imperfecta: An expanding panorama of variants. Clin Orthop Res 1981;159:11-25.
3. Remigo PA, Grinvalsky HT. Osteogénesis imperfecta congénita. Am J Dis Child 1970;119:524.
4. Mc Kusick VA. Heritable disorders of connective tissue. 4 ed. St Louis: CV Mosby, 1972:390.
5. Jones KL. Smiths recognizable patterns of human malformation. 4 ed California: WB. Saunders, 1988:436-7.
6. Shapin JE, Phillips JA, Byers PH. Prenatal diagnosis of lethal perinatal osteogenesis imperfecta (OI Type II). J Pediatr 1982;100:127-33.
7. Pal A, Boleman I, Kovacs D. A magzati femur hosszmérésének jeletösége. Orv Hetil 1995;136(44):2399-400.
8. Berge LN, Marton V, Tranebjaerg L. Prenatal diagnosis of osteogenesis imperfecta. Acta Obstet Gynecol Scand 1995;74(4):321-3.
9. Chalubinski K, Plenk H Jr, Schaller A. Prenatal diagnosis of osteogenesis imperfecta. Report of a case classified as the classical vrolik lethal type. Ultraschall Med 1995;16(1):25-8.
10. Herrera M, Ruiz M, González P. Osteogénesis imperfecta: prenatal diagnosis. Rev Chil Obstet Ginecol 1989;54(1):26-33.
11. Cole WG, Dalgleish R. Perinatal lethal osteogenesis imperfecta. J Med Gen 1995;32(4):284-9.
12. Verkh Z, Ruseell M, Miller CA. Osteogénesis imperfecta type II: microvascular changes in the CNS. Clin Neuropathol 1995;14(3):154-8.
13. Raghunath M, Makay K, Dalgleish R. Genetic counselling on brittle grounds: recurring osteogenesis imperfecta due to parental mosicism for a dominant mutation. Eur J Pediatr 1995;154(2):123-9.
Recibido: 2 de noviembre de 1998. Aprobado: 26 de noviembre de 1998.
Dr. Ulises D. Lima Rodríguez. Edificio 55, apartamento 7, Micro "A", Ciego de Ávila. Cuba. CP 65200.