Características clínicas y genéticas del Síndrome de Usher
Clinical Characteristics of the Usher's syndrome
Dra. Elisa Dyce Gordon; Yolanda Mapolón Arcendor: Dra. Maritza Palma López; Dr. C Jorge Santana Alvarez
Centro de Retinosis Pigmentaria de Camagüey. Camagüey, Cuba.
]]>
RESUMEN
Con el objetivo de describir las características clínicas y genéticas del síndrome de Usher, se realizó un estudio descriptivo transversal en el Centro de Retinosis Pigmentaria de Camagüey, desde octubre de 1991 hasta Mayo de 1998, con siete pacientes con síndrome Usher. A través de entrevistas y confección de encuestas, incluyendo el árbol genealógico se recopilaron los datos necesarios; los cuales fueron procesados realizándose estadística descriptiva. El síndrome de Usher tipo II fue el más frecuente. El inicio de la hipoacusia y de las manifestaciones oftalmológicas más frecuentes fueron la ceguera nocturna y la fotofobia. Hubo consanguinidad en el 28, 57 % de las familias y los antecedentes patológicos se presentaron en tres familias (43%). La enfermedad es clínica y genéticamente heterogénea. Debe ser sospechada en todo paciente con retinosis pigmentaria e hipoacusia. El diagnóstico debe realizarse precozmente.
DeCS: SÍNDROME USHER; RETINOSIS PIGMENTARIA; SORDERA.
ABSTRACT
State the clinical and genetic characteristics of the Usher syndrome, a descriptive and transversal study was carried out at the Camagüey retinosis pigmentaria Center, since octuber 1991 until may 1998 with seven patients affected by Usher syndrome. By interviews and preparation of surveys, including the genealogical tree necessary data were collected. Esher syndrome type II was the most frequent. Hearing loss and ocular manifestations onset were variable. Nigth blindness and phoptofobia were to the more frequent of the ophptalmologic manifestations. Consaguinity within families and pathological antecedents were found in 28, 57% and 43% respectively. The disease is clinical and genetically heterogenous. It must he suspected in all patients with retinitis pigmentosa and hearing loss. The diagnosis may be done early.
DeCS: USHER SYNDROME; RETINITIS PIGMENTOSA; DEATINESS.
INTRODUCCIÓN
]]> El síndrome de Usher (SU) es una enfermedad caracterizada por sordera sensorineural y trastornos visuales secundarios a retinopatía pigmentaria progresiva (retinosis pigmentaria (RP); descrita por el Dr. Charles Usher en 1914, quien también enfatizó su naturaleza hereditaria.1,2 No aparece con frecuencia, sin embargo, actualmente es la primera causa de limitación visual -auditiva o sordo-ceguera3 y es la forma sindrómica de la RP más frecuente.4,5 Debido a que en nuestro país se lleva a cabo un Programa de atención a la RP que incluye entre sus objetivos la caracterización genético-clínica de la RP, tanto en forma típica, atípica como sindrómica5 nos motivamos a realizar el presente trabajo cuyo objetivo es describir las características clínicas y genéticas del SU en nuestro medio.
MÉTODO
Se realizó estudio descriptivo transversal con siete personas que padecen el síndrome de Usher (SU), diagnosticadas desde octubre de 1991 hasta mayo de 1998 en el centro de Retinosis Pigmentaria (RP) de Camagüey.
Todos los pacientes acudieron al centro por problemas visuales. Se les realizó examen físico, perimetría y electrorretinograma. Posteriormente, debido a la presencia de hipoacusia fueron remitidos a la consulta de otorrinolaringología donde se realizó el diagnóstico de Síndrome de Usher a través de examen físico, pruebas audiológicas y vestibulares, además fueron clasificados.
Todos los pacientes fueron entrevistados por el genetista. Se les confeccionó una encuesta en la cual fueron vertidos los siguientes datos: clasificación del síndrome, edad de inicio de la hipoacusia y de los problemas visuales, así como los principales síntomas oftalmológicos y edad al diagnóstico.
La aparición de las manifestaciones oftalmológicas fue clasificada según la escala cubana de retinosis pigmentaria: precoz (inicio con menos de 10 años), juvenil (inicio entre 10 y 20 años).6 Se incluyó además la confección del árbol genealógico donde puede observarse la presencia o no de consanguinidad y los antecedentes patológicos familiares. Se les ofreció asesoramiento genético, orientándose la valoración oftalmológica y audiológica a los miembros de la familia con mayor riesgo, si ellos lo deseaban.
Los datos fueron procesados estadísticamente (estadística descriptiva), los resultados se presentan en tablas y gráficos explicativos.
RESULTADOS
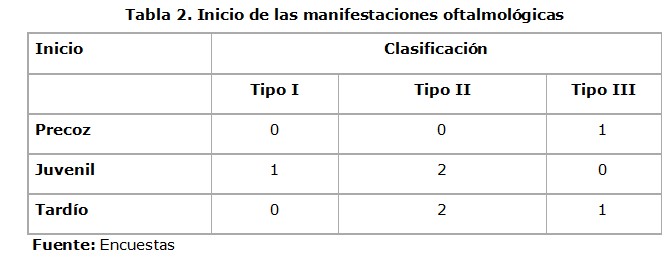
]]> Se encontró una mayor frecuencia del SU tipo II en el 57%, le siguieron en orden de frecuencia el tipo III con 29% y el tipo I con 14%.Las manifestaciones clínicas oftalmológicas tendieron al inicio juvenil y tardío (Tabla 2).
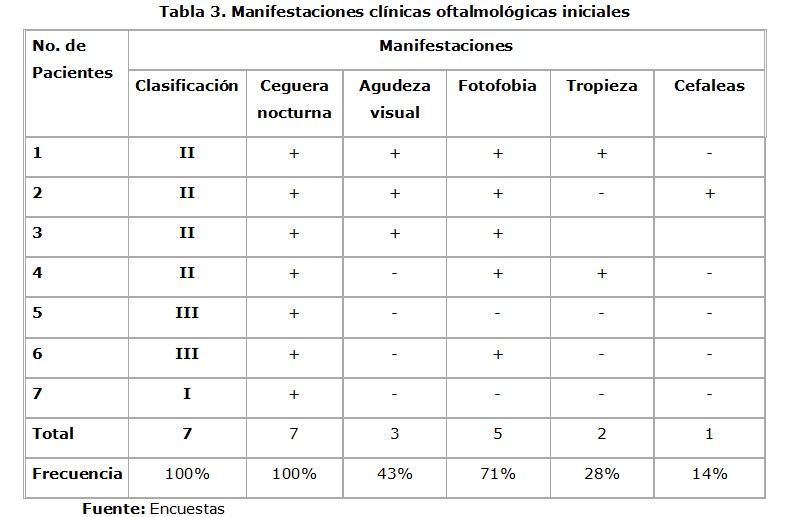
Las manifestaciones más frecuentes fueron la ceguera nocturna y la fotofobia, observándose que en los casos de SU tipo II los síntomas iniciales fueron numerosos (Tabla 3).
Hubo consanguinidad entre los padres de los afectados en el 28, 57% de los casos (N=2), no así en el 71, 4%. Los antecedentes familiares de SU, RP, o solamente hipoacusia se observaron en tres familias (42, 9%). El diagnóstico del SU en todos los casos fue realizado durante la adultez, como fruto del Programa Nacional de Atención de la Retinosis, ya que estos pacientes eran considerados como hipoacúsicos con problemas visuales o RP asociadas a hipoacusia.
DISCUSIÓN
Según la clasificación de Kimberling y Möller en 1995, el SU se clasifica en tres tipos (Usher I, Usher II y Usher III) (5) que difieren clínicamente, ya que cada uno tiene sus propias características. El SU tipo I se caracteriza por sordera sensorineural de profunda a severa, congénita, bilateral y simétrica, no progresiva. Inicio de la RP entre los ocho y los 15 años de edad, respuesta vestibular anormal.2,3 El SU tipo II presenta sordera sensorineural leve, moderada leve, moderada o severa, congénita bilateral, simétrica, no progresiva. Inicio de la RP después de los 15 años de edad, y respuesta vestibular normal o anormal.3
Esta homogeneidad clínica está determinada por la heterogeneidad genética, ya que cada uno de estos tipos de SU es causado por genes diferentes, localizados en distintos cromosomas que permiten la clasificación de cada tipo en subtipos, como se expone a continuación:
]]> TIPOS SUBTIPOS LOCALIZACION DEL GENUsher I Usher Ia 14q32
Usher Ib 11q13.5
Usher Ic 11p15.1
Usher Id 10q
Usher Ie 21q21
Usher II Usher IIa 1q41
Usher IIb not 1q
Usher III - 3q21-q25 7
En el presente estudio se evidencia esta heterogeneidad clínica y genética, al presentarse los tres tipos clínicos de la enfermedad. Aunque en otros países se reporta una mayor frecuencia del SU tipo I, en Cuba existe un predominio del SU tipo II sobre el tipo I,8 acorde con nuestros resultados.
]]> En los tres tipos descritos, el inicio de las manifestaciones oftalmológicas y trastornos vestibulares coinciden con los reportes de la literatura.1-3 En cuanto al inicio de la hipoacusia, en los pacientes con SU tipo II a pesar de que ésta es congénita, ellos refieren un comienzo durante la adolescencia. El hecho de ser esta hipoacusia menos severa, que permite un desarrollo adecuado del lenguaje, justifica que los pacientes no se hayan percatado de la misma hasta llegar a esta etapa.Acorde con el patrón de herencia autosómico recesivo, los padres y el 50% de los hermanos de los pacientes con el SU son portadores y estos pueden tener discretas alteraciones auditivas y oftalmológicas.
Para realizar detección de portadores sería conveniente realizar electrorretinograma y audiometría a los mismos, aunque algunos investigadores consideran que los métodos audiométricos no son útiles para identificar portadores, fundamentalmente en el SU tipo I (9); por lo tanto, hasta el momento el único método certero de detección de portadores es el estudio molecular.
CONCLUSIÓN
Esta enfermedad es clínicamente heterogénea. Debido a la combinación de ceguera y sordera, que tiende a limitar severamente a estos pacientes, y a su naturaleza hereditaria, se requiere del diagnóstico precoz para comenzar tratamiento adecuado, orientar la educación de estos individuos y para evitarles accidentes; así como para ofrecer asesoramiento genético a los padres y así ayudarlos a tener una actitud adecuada en cuanto a planificación familiar.
REFERENCIAS BIBLIOGRAFICAS
1. Kimberling W, Smith RJH. Gene mapping of the Usher syndromes. Otolaryngol North Am. 1992; 25(5): 923-34.
]]>2. Kimberling W, Möller C. Clinical and molecular genetics of Usher syndrome. J Am Acad Audiol. 1995; 6(1): 63-72.
3. Tamayo ML. La sordo-ceguera: un reto actual. En: Tamayo ML. Manual de genética de la retinosis pigmentaria.y el síndrome de Usher.. Bogotá, Colombia: INCI; 1996: 25-31.
4. Ayuso C, Antiñolo G. Retinitis pigmentosa. Clin genet. 1995;48(3):120-2.
5. Sarmiento JA. Algunas variaciones epidemiológicas de la retinosis pigmentaria en Cuba. En: Peláez O. Retinosis Pigmentaria. Experiencia cubana. La Habana: Editorial Científico técnica; 1997.p.35-47.
6. Herrera M. Clasificación. En: Peláez O. Retinosis Pigmentaria. Experiencia cubana. La Habana: Editorial Científico técnica; 1997.p.67-82.
]]>7. MacDonald IM, Haney PM, Musarella MA. Summay of ocular genetic disorders and inherited systemic conditions with eye findings. Ophtalmic Genet. 1998;19(1):1-17.
8. Herrera M. Características Clínicas de la Retinosis Pigmentaria. En: Peláez O. Retinosis Pigmentaria. Experiencia Cubana. La Habana: Editorial Científico -Técnica; 1997.p. 87-107.
9. Wagenaar M, Smik AF. Carriers of the Usher syndrome type Ib : is audiometric identification possible?. Am J Otol. 1996;17(6):853-58.
Dra. Elisa Dyce Gordon. Especialista de II Grado en Genética Clínica. Profesora Asistente. Centro de Retinosis Pigmentaria de Camagüey. Camagüey, Cuba.
]]>{kind=link}
{kind=link}