Síndrome de Alport: herencia y trasplante renal en una familia
Alport’s ´syndrome: heritage and kidney transplantation in a family
Dr. Rafael Pila Pérez; Dr. Miguel Rivero Sánchez; Dr. Rafael Pila Peláez; Dr. Francisco Ávila Riopedre
Hospital Clínico Quirúrgico Docente Manuel Ascunce Domenech. Camagüey, Cuba.
]]>
RESUMEN
Se trata de una familia distribuida en tres generaciones afectada por nefropatía familiar hematúrica (Síndrome de Alport-Perkoff) En tres de ellos se asoció la nefropatía y la sordera, además padecieron de insuficiencia renal crónica y fueron transplantados. Uno de ellos falleció por linfoma primario cerebral y los otros dos no presentaron alteraciones de la función renal; en seis de ellos se constató hipoacusia, mientras que un solo paciente, el más joven de esta familia, no mostró síntomas. Las mujeres presentaron trastornos en etapas más avanzadas de la vida. Se señalan las características hereditarias, clínicas y terapéuticas de esta enfermedad.
DeCS: NEFRITIS HEREDITARIA; TRANSPLANTE DE RIÑÓN; LINAJE.
ABSTRACT
This work deals with a family distributed in three generations affected by family hematuric nephropaty (Syndrome of Alport-Perkoff). In three of them, nephropaty and deafness are associated, they sufferred for chronic renal failure and were trasplanted. One of them died for primary brain lymphoma and the other two did not present renal function disorders; in six of them it was detected hypoacoustics, while one patient (the younger) of this family was free of symptoms. We observed that women presented disorders in more advanced stages of life. Hereditary; clinic and therapeutic characteristics are pointed out.
DeCS: HEREDARY NEPPHRITIS; KIDNEY TRANSPLANTATION; PEDIGREE.
INTRODUCCIÓN
]]> Esta enfermedad se caracteriza por la presencia de una nefropatía en la que el síntoma cardinal es una hematuria persistente asociada a una sordera neurógena y con menos frecuencia alteraciones del tipo oftálmico.Este síndrome es conocido hace casi un siglo en que Dickinson 1 lo describe por primera vez en una familia de 17 miembros, pero es Alport, 2 y más tarde Perkoff, 3 quienes describieron detalladamente este síndrome, observaron la constante asociación de la sordera a la nefropatía y la gravedad clínica que está presente en el varón y sobre todo ciertas características en cuanto al tipo de transmisión hereditaria.
Aún no existe un criterio unánime en cuanto a la etiopatogenia de esta enfermedad. Consiste en una nefropatía evolutiva hacia la insuficiencia renal crónica (IRC), más grave para los varones con sordera de percepción, habitualmente bilateral, especialmente para las altas frecuencias. Las mujeres suelen ser muchas veces asintomáticas. 4, 5
Se ha señalado como rasgo característico de esta afección la presencia de otitis, 4 también se han descrito alteraciones oftalmológicas como cataratas, lentícono, nistagmos, retinitis pigmentaria, miopía, microesferofaquia, 5 asimismo aminoaciduria, polineuritis, espina bífida, cardiopatías, alteraciones de los lípidos, de las plaquetas y alteraciones cerebrales.
El objetivo de este trabajo es exponer el estudio de una familia, analizar la clínica y mostrar tres pacientes que llegaron al trasplante renal por IRC debido a esta enfermedad.
Estudio familiar
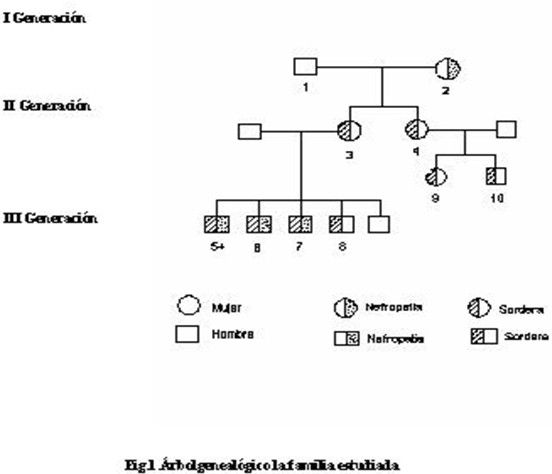
En relación con el esquema de distribución de las tres generaciones conocidas de la familia estudiada (Figura 1), de la primera sólo se conocen detalles clínicos poco precisos de la abuela de los pacientes estudiados, que padeció de hipertensión arterial (HTA) y de sordera en edades tempranas de la vida, no pudo ser estudiada porque falleció.
En la segunda generación los dos hijos del matrimonio presentaban hipoacusia de percepción y además HTA, no mostraron daño renal, sólo uno de ellos trastornos visuales provocados por cataratas bilaterales.
De los siete hijos resultantes de los dos matrimonios de la segunda generación, seis presentaron hipoacusia de carácter neurógeno, tres de ellos se acompañaban de nefropatía grave que los llevó a la IRC y al trasplante renal, uno de ellos falleció por linfoma primario del cerebro. EL enfermo más joven de este estudio se encontraba asintomático. (Figura 1)
]]> Caso clínico 1 (No. 5 del árbol genealógico)Exámenes de laboratorio: discreta anemia y elevación de la velocidad de eritrosedimentación (VSG). Creatinina: 220 mmol/l; glicemia: normal.
Proteinograma: Elevación de las globulinas alfa y gamma con discreta disminución de las proteínas totales.
Estudio lipídico: elevación del colesterol, triglicéridos y lípidos totales.
Electrolitemia: hiponatremia e hiperfosfatemia.
Electrocardiograma (ECG): HVI, electroencefalograma (EEG): normal.
Conteo de Addis y orinas parciales: con predominio de leucocituria, hematuria y proteinuria discreta.
Fue dado de alta con el diagnóstico de Síndrome de Alport y posteriormente ingresó en múltiples ocasiones con la misma afección. Varios años después se detectó sordera de carácter absoluto de percepción bilateral y en otro ingreso se encontraron alteraciones visuales en fondo de ojo, entre ellas la retinitis pigmentaria. Se atendió por nefrología ya que desde los 12 años presentaba IRC y requirió tratamiento con hemodiálisis.
Al cumplir los 15 años fue atendido en nuestro hospital, sometido a diálisis y hemodiálisis por estar en estadio IV de IRC. En 1992, con 19 años de edad, se le realizó trasplante renal. Seis años después falleció por linfoma primario de cerebro, pero con función renal normal.
Estudios de laboratorio: Hb: 9,9 g/l; leuc: 5800 x 109; diferencial: normal; eritrosedimentación: 110 mm/h; creatinina: 800 ml/l; glicemia: normal; colesterol, triglicéridos y lípidos totales: elevados. Proteinograma: normal. Calcio y fósforo: normales. Sodio: hiponatremia. Proteinuria: moderada. Coagulación y plaquetas: normales. ECG: taquicardia sinusal. EEG: normal. Audiometría: sordera de percepción bilateral endolaberíntica. F.O.: cataratas bilaterales, lentícono. Imagenología de tórax, cráneo, survey óseo: normal. USG: riñones pequeños de contornos irregulares sin pielocalectasias.
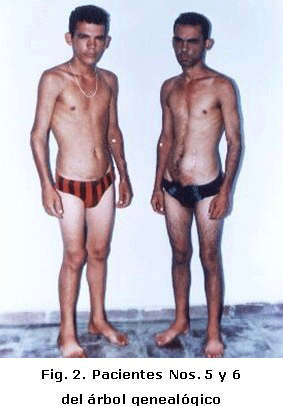
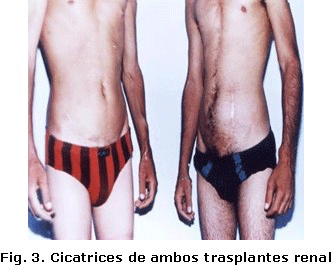
Fue tratado por varios años por nefrología en el Hospital Pediátrico; al llegar a los 15 años se remitió a nuestro hospital con cifras de creatinina de 2 110 mmol/l, filtrado glomerular de IRC en estadio IV, por lo que el plan dialítico no resolvió su enfermedad y fue preparado para trasplante renal, el cual se realizó el 4 de mayo de 1994, tuvo tratamiento con drogas para el trasplante y la HTA, posteriormente mantuvo la normofunción renal. Recibió seguimiento cada dos meses por nefrología. El paciente actualmente tiene 27 años. Figuras 2 y 3.
Caso 3 LNG (No. 7 del árbol genealógico)
Varón de seis años de edad que fue ingresado en el Hospital Pediátrico con una gromerulopatía primaria. Al conocer el antecedente familiar fue ingresado en varias ocasiones por Síndrome de Alport, con antecedentes de amigdalitis y otitis a repetición. ]]>
Refiere que después de realizar ejercicios físicos aparecieron orinas oscuras como agua de lavado de carne alternadas, en ocasiones, con orina de aspecto normal.
En uno de los ingresos se constató sordera bilateral con predominio en el oído izquierdo y miopía bilateral, por lo que necesitó usar lentes. También presentó disnea a los esfuerzos, polaquiuria y tenesmo vesical.
Examen físico: Corazón: SS II/VI en todos los focos de auscultación.
Estudio analítico: Hb: 8,8 gr/l, leuc.: 6000 x 109. Diferencial: normal. VSG: 60 mm/h. Creatinina: 560mmol/l. Lipidograma con colesterol, triglicéridos y lípidos totales: elevados. Proteínas totales: 6,95 g/100. Coagulación y plaquetas: normales. EEG: normal. Audiometría: sordera de percepción bilateral endolaberíntica. Exploración oftalmológica: miopía con retinosis miopática. Radiografía de tórax, cráneo y súrvey óseo: normales. Ecografía abdominal: riñones pequeños sin otras alteraciones.
El paciente fue atendido por nefrología por padecer IRC en el Hospital Pediátrico y fue referido a nuestro hospital donde se preparó para realizarle trasplante renal por métodos dialíticos. El tratamiento se efectuó en marzo de 1998, el enfermo hizo rechazo agudo no mediado por anticuerpos preformados, aunque logró aceptarlo a los 90. Desde ese momento la función renal fue normal. Figuras 2 y 3.
Caso 4 (No. 8 del árbol genealógico)
Varón de 16 años de edad, fue atendido por consulta externa porque presentó hipoacusia endolaberíntica bilateral, sin alteraciones renales.
Exámenes de laboratorio: normales.
Caso 5 (No. 9 del árbol genealógico)
Varón de 14 años, sin alteraciones. ]]>
La hembra No. 4 de la segunda generación tuvo dos hijos, una hembra y un varón, sólo se constató hipoacusia bilateral en el varón e hipoacusia del oído izquierdo en la hembra, sin daño renal u otra afección en estos momentos.
COMENTARIOS
El Síndrome de Alport es un trastorno hereditario que se caracteriza por hematuria .En la actualidad se reconocen cuatro formas:
a) La clásica, que es un trastorno ligado al cromosoma X caracterizado por hematuria, sordera sensitivo-neural y deformación crónica de la superficie anterior del cristalino (lentícono).
b) Una segunda forma ligada al cromosoma X que se asocia a leiomiomatosis difusa.
c) Forma autosómica recesiva.
d) Una forma autosómica dominante.
Las dos formas autosómicas pueden causar enfermedad renal sin sordera ni lentícono, la incidencia de este síndrome es de alrededor de 1 por 10 000 en la población general. 5, 6
Esta enfermedad hereditaria está ligada al cromosoma X y presenta mutaciones en los genes para las cadenas α2, α4, α5 y α6 del colágeno de tipo IV, un componente fundamental de las membranas basales. 6 Se caracteriza por insuficiencia renal progresiva de gravedad variable, es más grave en los varones, provoca sordera neurosensorial para las altas frecuencias y anomalías oculares; 7 además, es la más frecuente de las enfermedades renales hereditarias. 8 ]]> Esta enfermedad es de larga duración, se establece durante años como se aprecia en nuestros pacientes, pero se ha observado también en los primeros meses de la vida.
Los síntomas que han predominado en estos enfermos son la hematuria, sordera y manifestaciones oculares. La hematuria puede ser variable desde varios miles de elementos por minuto hasta una franca hematuria macroscópica, con frecuencia puede ser aislada, pero se asocia a una proteinuria o a una leucocituria como se aprecia en estos enfermos. No es raro que esta enfermedad se presente de entrada, sobre todo en el varón, 9 acompañada de una IRC más o menos grave, como ocurrió con nuestros pacientes que necesitaron de trasplante renal. En la mujer el curso puede permanecer latente durante años o manifiestarse con pequeñas hematurias. La afección es más grave en varones que en hembras y en los primeros se desarrolla la IRC en la segunda y tercera décadas de vida. En la familia objeto de estudio se manifestó de esa forma. La proteinuria puede estar ausente o ser ligera al principio, pero con el tiempo el 75 % de los casos presentan algún grado de proteinuria. Los enfermos estudiados presentaron edemas, mantuvieron proteinuria con lípidos elevados, lo que se relaciona con un síndrome nefrótico, pero siempre se ha considerado muy raro. 10
La sordera suele ser precoz y grave, sobre todo en el varón, se acompaña de nefropatía y puede permanecer aislada como se apreció en esta familia, e incluso faltar en alguna generación. Es una sordera de tipo neurógena por afectación del nervio auditivo o del órgano de Corti, la lesión suele ser bilateral, aunque en ocasiones se presenta de un solo lado. Se encuentran afectadas las frecuencias 4096 – 2048 y más tardíamente, la 1024.
Por otro lado, la otitis es señalada por algunos autores 5, 6 como un rasgo bastante característico, quizás como traducción de la facilidad que presentan para las infecciones focales. Todas estas manifestaciones se comprobaron en nuestros pacientes, se observó que puede existir enfermedad renal grave con sordera y sordera sin afectación renal.
Los síntomas oculares no son típicos, aunque sí frecuentes. Los más comunes son la catarata, miopía, lentícono, retinitis pigmentaria, microesferofaquia e hipermetropía. 11 Nuestros pacientes presentaron fundamentalmente los tres primeros.
Este síndrome y sus variantes son enfermedades que consisten en un trastorno del metabolismo de la membrana basal glomerular que en la nefritis hereditaria se manifiesta por una reduplicación 12 y en el síndrome de hematuria recidivante, por una membrana basal muy fina que constituyen sus diagnósticos diferenciales más importantes. 12, 5, 6 Las células espumosas intersticiales consideradas por algunos autores 12, 13 si no específicas, sí bastante características, se observan aproximadamente en una tercera parte de los pacientes, principalmente en la unión corticomedular.
No se sabe si la herencia está ligada al cromosoma X o si es autosómica con una penetrancia importante en el cromosoma X, 14 por lo que la enfermedad suele ser grave y se manifiesta con más frecuencia en los varones, como lo reportamos en esta serie.
Tampoco existe terapia específica; 15 las medidas terapéuticas comunes de la IRC y sus complicaciones deben utilizarse cuando se presenten, mientras que la diálisis o el trasplante renal deben efectuarse cuando sobrevenga la IRC avanzada. Tres de nuestros enfermos fueron asistidos de esta forma hasta el trasplante, el caso No. 5 falleció con función renal normal después de seis años, mientras que los casos Nos. 6 y 7 se encuentran con normofunción renal después de ocho y siete años del trasplante renal, respectivamente.
REFERENCIAS BIBLIOGRÁFICAS
]]>1. Dickinson W. Disease of the kidney and urinary derangement. Longmas Green: Ed. London; 1965. p. 130-7.
2. Alport A. Hereditary familial congenital haemorrhagic nephritis. Brit Med J. 1927; 1:504-8.
3. Perkoff G. Familial aspects of diffuse renal disease. Ann Rev Med. 1964; 25(5):115-9.
4. Crawford M, Toghill P. Alports Syndrome of hereditary nephritis and deafness and ocular lesions. Quart J Med. 1998;7: 3-70.
5. Omeara Y, Brady H, Brenner B. Glomerulopatías asociadas a las enfermedades multisistémicas. En: Fanci A, Braunwald E, Isselbach K, Martin J, editors. Harrison: principios de Medicina Interna. 14 ed. México: Ed McGraw-Hill; 2000. p. 1755-64.
]]>6. Martínez Maldonado M. Nefropatías crónicas hereditarias. En: Bennett J, Plum F, editors. Cecil: principios de Medicina Interna. 20 ed. La Habana: McGraw-Hill Interamericana; 2000. p. 697-702.
7. Callis L. Nefropatía hematúrica familiar de Síndrome de Alport. Rev Española Ped. 1997;53: 83-9.
8. Gluber M, Levy M, Broker M. Alports Syndrome: a report of 58 cases and a review of the literature. Am J Med. 1991;80: 493-8.
9. Valderrabano F. Varón de 4 años en uremia por nefropatía familiar no filiada e insuficiencia aórtica corregida con prótesis. Sesión clínicopatológica. Rev Clin Esp. 1997; 51:6-20.
10. Grunfeld J. The clinical spectrum of hereditary nephritis. Kidney Int. 1997; 39:83-92.
]]>11. Ohlsson L. Congenital renal disease deafness and myopia in one family. Acta Med Scand. 1997;198:77-83.
12. Behrman R, Vaughan V, Nelson N. Enfermedades hereditarias o familiares. En: Behrman R, Vaughan V, Nelson W, editors. Tratado de Pediatría. 10 ed. México: Ed McGraw-Hill Interamericana; 1994. p. 1397-99.
13. Cohen H, Hoyer J. Nephronothisis. a primary tubular basement membrana defect. Lab Invest. 1998;67:564-72.
14. Coles G, Robinson K, Branch R. Familial insterticial nephritis. Clin Nephrol. 1997;27:513-7.
]]> Recibido: 3 de noviembre de 2003
Aceptado: 17 de febrero de 2004
Dr. Rafael Pila Pérez.Especialista de II Grado en Medicina Interna. Profesor Titular del ISCM de Camagüey. Hospital Clínico Quirúrgico Docente. Manuel Ascunce Domenech. Camagüey, Cuba.
]]>