CASOS CLÍNICOS
Neurofibromatosis segmentaria: presentación de un caso
Segmentaryneurofibromatosis: a case presentation
Dra. Idalmis Campollo RodríguezI; Dr. José Luis Rodríguez RojasII; Dra. Leonor Maribel Limache YaringañoIII
I Especialista de I Grado en Dermatología. Profesor Instructor. Hospital Provincial Universitario Amalia Simoni. Camagüey, Cuba.icr@finlay.cmw.sld.cu. ]]>
II Especialista de I Grado en Dermatología. Máster en Medicina Natural y Tradicional. Profesor Instructor. Hospital Provincial Universitario Amalia Simoni. Camagüey, Cuba.
RESUMEN
Fundamento: las neurofibromatosis constituyen un grupo de enfermedades neurocutáneas, de herencia autosómica dominante que muestran extrema heterogeneidad clínica y están caracterizadas por crecimiento anormales en tejidos derivados de la cresta neural embriogénica afectando por tanto la piel, tejidos blandos, sistema nervioso y hueso. Objetivo: reportar un caso de neurofibromatosis segmentaria. Caso Clínico: paciente femenina de 42 años de edad de piel blanca con antecedentes de salud, que presentó una neurofibromatosis segmentaria con neurofibromas como manifestación única de la enfermedad.Conclusiones: la neurofibromatosis segmentaria es una enfermedad poco frecuente y de difícil manejo debido a la alta tasa de recidiva de los neurofibromas. El consejo genético es muy importante, como en el resto de las neurofibromatosis. Si los pacientes presentan mosaicismo gonadal pueden tener descendencia con NF1 clásica. Los enfermos con este tipo de NF tienen menor riesgo de desarrollar tumores malignos de la vaina periférica, pero aun así se recomienda seguimiento.
DeCS: NEUROFIBROMATOSIS 1; CRESTA NEURAL; MOSAICISMO; ADULTO; ESTUDIOS DE CASOS.
ABSTRACT
Background: neurofibromatosis constitute a group of neurocutaneous diseases, of dominant autosomal heredity that show extreme clinical heterogeneity and characterized by abnormal growth of tissues derived from the embryogenic neural crest, affecting therefore the skin, soft tissues, nervous system and bone. Objective: to report a case of segmental neurofibromatosis. Clinical case: a 42 years old, white skin, female patient with health antecedents, which presented a segmental neurofibromatosis with neurofibroma as unique manifestation of the disease. Conclusions: segmental neurofibromatosis is an infrequent disease and difficult to handle due to the high recidivation rate of neurofibroma. Genetic counseling is very important, as in the rest of neurofibromatosis. If patients have gonadal mosaicism may have offspring with classical NF1. Patients with this type of NF have lower risk of developing malignant tumors of peripheral sheath, but follow-up is still recommended.
DeSC: NEUROFIBROMATOSIS 1; NEURAL CREST; MOSAICISM; ADULT; CASE STUDIES.
INTRODUCCIÓN
Las neurofibromatosis constituyen un grupo de enfermedades neurocutáneas, de herencia autosómica dominante que muestran extrema heterogeneidad clínica. Se caracterizan por crecimientos anormales en tejidos derivados de la cresta neural embriogénica que afectan la piel, los tejidos blandos, el sistema nervioso y los huesos. 1
Según Riccardi, citado por Fuente Rodríguez, 2 existen ocho grupos de neurofibromatosis (NF) que se clasifican en relación a las manifestaciones clínicas. La forma más frecuente es la NF tipo I o enfermedad de Von-Recklinghausen que incluye más del 85 % de los casos. Los pacientes tienen muchos neurofibromas que miden desde escasos milímetros a pocos centímetros de diámetro, numerosas manchas de color café con leche ampliamente distribuida, escasa o ninguna lesión en el sistema nervioso central. Los nódulos de Lysch pueden encontrarse en los iris de aproximadamente un cuarto de los pacientes por encima de los seis años de edad y en el 94 % de enfermos con mayor edad. El tipo II, NF central o acústica (Schwanomaacústico), se distingue por neurinoma del acústico bilaterales, pocas manchas color café con leche y ausencia de nódulos de Lysch. El tipo 3 (mixto) y 4 (variante) recuerdan al tipo 2, pero pueden tener numerosos neurofibromas cutáneos. Los pacientes que presentan esta enfermedad tienen un gran riesgo de desarrollar gliomas ópticos, neurilemomas y meningiomas.
Este tipo de formas se heredan como rasgos autosómicos dominantes. El tipo 5, NF segmentaria (dermatomal), se considera que surge por una mutación somática poscigótica y generalmente no es heredable. Se caracteriza por limitación de manchas color café con leche y/o neurofibromas en un segmento unilateral aunque puede ser bilateral. La NF tipo 6 no tiene neurofibromas, sólo manchas color café con leche, y debe darse en dos generaciones para poder ser diagnosticada. La NF tipo 7, neurofibromatosis de inicio tardío, no cursa con neurofibromas hasta que los enfermos alcanzan la segunda década y aún no se conoce si es hereditaria. La NF tipo 8 o no especificado, incluye a los pacientes que presenten una neurofibromatosis definida, no es característica de otras categorías. 3, 4 En la actualidad se definen dentro de las neurofibromatosis los siguientes síndromes: NF tipo 1 o enfermedad de Von-Recklinghausen, NF tipo 2, NF tipo 1 segmentaria o en mosaico, manchas color café con leche familiares y schwanomatosis. 1
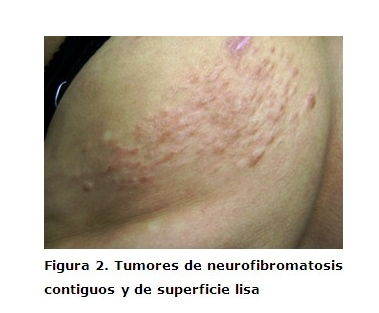
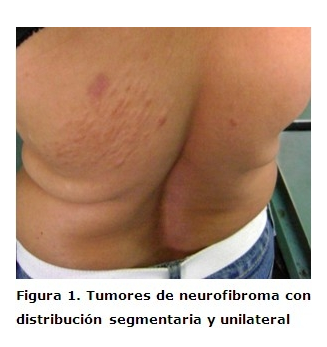
Las primeras referencias acerca de la neurofibromatosis segmentaria (NFS) citadas por Saettone León, 5 fueron hechas por Gammel en 1931. Dos décadas más tarde, Miller y Sparkers citados por el propio autor proponen el término de NFS. La NFS es una forma de presentación de NF tipo1 que se caracteriza por neurofibromas y manchas color café con leche o sólo neurofibromas, confinados a un territorio circunscripto en el cuerpo, o sea con distribución segmentaria y unilateral. Es un trastorno raro cuya afección es 10 a 20 % menos frecuente que en la NF tipo I. Su prevalencia ha sido estimada en 1 cada 40.000 individuos de la población general. 6
Este trabajo presenta a un paciente con NFS con neurofibromas como manifestación única de la enfermedad.
CASO CLÍNICO ]]>
Paciente femenina de 42 años de edad, color de la piel blanca; la misma acudió al servicio de Dermatología del Hospital Universitario Amalia Simoni de Camagüey por presentar varias lesiones tumorales de aproximadamente 0.5mm de diámetro aplanados y sésiles del color normal de la piel, no dolorosos, los cuales comenzaron a aparecer en la adolescencia y fueron aumentando en número y tamaño con el transcurso de los años, localizados en el tórax izquierdo seguido de una distribución neural en dermatomas. Tienen una consistencia blanda y a la compresión tienden a invaginarse a través de un pequeño orificio en la piel (signo del ojal). Al examen físico no se encontraron manchas color café con leche, pecas, ni compromiso sistémico. (Figuras 1 y 2)No presenta antecedentes patológicos familiares de la enfermedad. Con estos datos clínicos se realizó el diagnóstico de neurofibromatosis segmentaria, ordenándose examen oftalmológico y biopsia de piel. El examen oftalmológico fue normal y el examen histopatológico fue compatible con neurofibroma. (Figura 3)
No se realizó un estudio genético sobre el tejido cutáneo afectado para buscar el mosaicismo somático, ni sobre el tejido gonadal, pues al no existir un tratamiento específico para los neurofibromas por su difícil control postquirúrgico y la alta tasa de recurrencia a largo plazo, se decidió mantener un seguimiento rutinario para la detección precoz de complicaciones ya que son considerados benignos, aunque se encuentran descritos en la literatura pacientes a los que se les ha desarollado tumores malignos de nervios periféricos.
DISCUSIÓN
Saettone 5 cita que se atribuye a Von Tilesius la descripción de neurofibromatosis en 1793, pero, según datos de Sezer E, 7 el primero en describir lo que ahora se conoce como neurofibromatosis tipo 1 fue el médico, morfofisiólogo y botánio Rudolph Albert von Kölliker en 1860. González 6 cita que Friedrich Daniel Von Recklinghausen (1833-1910), médico y patólogo, elaboró la descripción clásica e incluyó fotografías. Fuente Rodríguez 2 refiere que durante el siglo XX destacó el papel fundamental del doctor Riccardi, médico encargado de la fundación Texana para la Neurofibromatosis, y que aportó muchos avances en el conocimiento de esta enfermedad. Mientras que Sezer 7 cita que sobre la neurofibromatosis segmentaria, los pioneros en describir las formas localizadas fueron Gammel en 1931 y Crowe en 1956 quienes además utilizaron el término neurofibromatosis sectorial.
Listernick 1 y Vargas Martínez 8 mencionan la clasificación de la NFS, revisada por Rothen en 1987, la cual comprende cuatro subtipos: la segmentaria verdadera o la forma clásica de Riccardi (unilateral, cutánea no hereditaria), la segmentaria con compromiso visceral profundo (no familiar), la segmentaria con casos familiares (sin compromiso profundo) y la segmentaria cutánea bilateral (sin compromiso profundo, no familiar). A su vez Landrie 9 y Terzi 10 hacen referencia a una nueva clasificación surgida en 1994, la cual tiene en cuenta la hipótesis de mutación somática. En la misma se describe que las características clínicas de la enfermedad estarían determinadas por el momento del desarrollo embriológico en el cual ocurre la mutación (precoz o tardía) y por la fuente de tejido afectado por la mutación. ]]>
La hipótesis de la mutación somática concluye que los pacientes con NF tipo 1, generalizada con mosaicismo a nivel molecular, presentarían mutaciones precoces, mientras que aquellos que desarrollan neurofibroma o manchas color café con leche aisladas, sin compromiso sistémico asociado (NFS), presentarían dicha mutación en células ya diferenciadas, en una etapa postcigóticas.Dado que el mosaicismo somático a consecuencia de una mutación producida posterior a la concepción, la NFS se considera esporádica aunque han sido descritos casos familiares.
Se conoce que el gen de la NF tipo 1 se encuentra en el cromosoma 17, el cual codifica un ARN de 60 exones, cuyo producto final es una proteína llamada neurofibromina, la cual estaría involucrada en el control del crecimiento y la diferenciación celular, con una acción tumoral supresora. La mutación postcigótica en el gen de la NF tipo 1 en las células derivadas de la cresta neural, se considera la base del mosaicismo genético, en el cual se pierde la neurofibromina en los fibroblastos y las células de Schwann, la misma determina la aparición de neurofibrosis o cambios pigmentarios respectivamente. 11
En la NFS con neurofibromas como manifestación única de la enfermedad, los NF siguen con una distribución neural en dermatomas, ya que la mutación genética se limita a las células de Schwann; mientras que aquellos con cambios pigmentarios realizan la mutación de los fibroblastos, y la distribución de las lesiones sigue las líneas cutáneas de Blaschko (no asociadas a estructuras nerviosas). 11, 12
Según Pascual Castroviejo, 13 la NFS debido a mosaicismo somático parece una variedad patológica de la NF tipo 1, que está infradiagnosticada y presenta tantas posibles localizaciones en el cuerpo como la NF tipo 1 sin mosaicismo. Además dice que la forma menos frecuente de presentación es la que sólo tiene manifestación cutánea.
Alrededor de 60 % de los pacientes con NFS es de sexo femenino. La edad de inicio varía desde el nacimiento hasta los 83 años, con una edad media de 28 años. La relación mujer / hombre es de 2:1, y presenta una incidencia en dos picos: 20 a 30 años y 50 años. Se caracteriza por manchas color café con leche, pecas y neurofibromas. Los sitios más afectados son: tórax y abdomen (55 %), brazos (20 %), miembros inferiores y cara (10 %). Por causa desconocida es más común del lado derecho que del izquierdo, aunque se han reportado pacientes con NF1 segmentaria bilateral. 8
Los neurofibromas en NFS son generalmente estructuras blandas, no dolorosas y raramente encapsuladas de difícil control postquirúrgico debido a la alta tasa de recurrencia a largo plazo. Los tumores que aumentan rápidamente de tamaño se deben extirpar inmediatamente puesto que pueden tornarse malignos
Las indicaciones para la exéresis quirúrgica de un neurofibroma incluyen dolor, compresión de estructuras adyacentes, desfiguración cosmética, deterioro neurológico y rápido crecimiento sugestivo de degeneración maligna. 14
CONCLUSIONES ]]>
La NFS es una enfermedad poco frecuente y de difícil manejo debido a la alta tasa de recidiva de los neurofibromas. Si los pacientes presentan mosaicismo gonadal pueden tener descendencia con NF 1 clásica, y a pesar de que los enfermos con este tipo de NF tienen menor riesgo de desarrollar tumores malignos de la vaina periférica, aún así se recomienda seguimiento.
REFERENCIAS BIBLIOGRÁFICAS
1. Listernick R, Charrow J. The neurofibromatoses. In: Wolff K, Goldsmith L, Katz SI. Fitzpatrick'sdermatology in general medicine. 7ed. Nueva York: McGraw-Hill; 2008. p. 1331-39.
2. Fuente Rodríguez N, Tápanes Domínguez A, Pérez La O P. Neurofibromatosis tipo 1, enfermedad de Von Recklinhausen. Rev Cub Med Mil (serie en internet). 2007 Dic [citado 2010 Abr 7]36(4):(aprox. 5 p). Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext pid=S0138-65572007000400009 & lng=es.
3. Kumar S, Nehal KS, Paller AS. Algunas genodermatosis y síndromes adquiridos. En: Odom R, James W, Berger T. Andrews' Dermatología clínica. Nueva York: Editorial Marbán; 2004: 682-92.
4. Ferner R. Neurofibromatosis 1 and neurofibromatosis 2: A twenty first century perspective. Lancet Neurol. 2007;6:340-51.
5. Saettone León A.Neurofibromatosis segmentaria, reporte de un caso. Dermatol Perú. 2006;16(1):249-53.
6. González G, Russi ME, Lodeiros A. Bilateral segmental neurofibromatosis: a case report and review. Pediatric Neurology. 2007;36(1):51-3.
7. Sezer E, Senayli A, Seazer T, Bicakci U. Segmental neurofibromatosis: Report of two cases. Journal of Dermatology. 2006;33(9):635-38.
8. Vargas Martínez F, Arenas R. Enfermedad de von Recklinghausen. Una visión actual de las neurofibromatosis.Dermatología Cosmética, Médica y Quirúrgica. 2009;7(3):181-89.
9. Landrie F, Ferrara P, Hem S, Carrizo A. Neurofibromatosis segmentaria frontotémporo orbitaria: Reporte de un caso. Revisión de la literatura. Rev Argent Neurocir. 2007;21(3):415-21.
10. Terzi Y, Oguzkan S, Aysun S, Ayter S. Neurofibromatosis: Novel and recurrent mutations in Turkish patients. Pediatr Neurol. 2007;37:421-25.
11. Jeblaoui Y, Neji B, Haddad S, Mnif D, Hchicha S. Difficults of themanagement of head and neck neurofibromatosis. Annales de Chirurgie Plastique et Esthetique. 2007;52(1):43-50.
12. Falcón Caballero J, Mantenga B, González García J. Neurofibromatosis, presentación atípica de un caso. Investigaciones Medicoquirúrgicas. 2007;1(9):56-60.
13. Pascual Castroviejo I, Pascual Pascual S, Velázquez Fragua R, Viaño J, López Gutiérrez J. Neurofibromatosis segmentaria en niños: presentación de 43 pacientes. Rev Neurol. 2008;47(8):399-403.
14. Pérez Hernández LM, Marrero Riverón LO, Roché Egues HE, Calzado Calderón R, Gonzálezde Varona C. Diagnóstico clínico imagenológico de la neurofibromatosis tipo 1. Rev Cub OrtopTraumatol. 2005;19(1):47-50.
Recibido: 7 de junio de 2010
Aprobado: 12 de febrero de 2011
Dra. Idalmis Campollo Rodríguez. Email: icr@finlay.cmw.sld.cu
]]>