Presentación de una familia con diagnóstico de encondromatosis múltiple familiar
Presentation of a Family with Multiple Family Enchondromatosis
Ana Iris Heres Zalazar 1, Arnolis Fuentes Rodríguez 2, Elayne Esther Santana Hernández 3, Orlando González Sale 4, Alina Fernández Oropesa 5
1. Máster en Asesoramiento Genético. Especialista de Primer Grado en Medicina General Integral. Instructor. Centro Municipal de Genética Cacocum. Holguín. Cuba.
2. Máster en Urgencias Médicas. Especialista de Primer Grado en Medicina General Integral. Especialista de Primer Grado en Gastroenterología. Instructor. Hospital Vladimir I Lenin. Holguín. Cuba.
3. Máster en Atención Integral al Niño. Especialista de Primer y Segundo Grado en Medicina General Integral y Genética Clínica. Asistente. Centro Provincial de Genética Médica de Holguín. Holguín. Cuba.
4. Licenciado en Biología. Instructor. Citogenetista del Centro Provincial de Genética de Holguín. Holguín. Cuba.
5. Máster en Asesoramiento Genético. Licenciada en Enfermería. Instructor. Centro Municipal de Genética Cacocum. Holguín. Cuba.
]]>
RESUMEN
La encondromatosis múltiple familiar fue descrita por Virchow en 1910, se presenta con un patrón de herencia autosómico dominante. Las encondromatosis comprenden un grupo heterogéneo de síndromes congénitos caracterizados por la presencia de encondromas múltiples, asociados a malformaciones músculos esqueléticos secundarios al acortamiento de las extremidades. Se presentó una familia con cuatro afectados, a los que se les realizó diagnóstico donde se aplicó el método clínico a través del examen físico e imagenológico. Es importante la realización de un diagnóstico precoz de esta entidad por la posibilidad de realizar una prevención secundaria en los pacientes, así como para brindar un adecuado asesoramiento genético a la familia.
Palabras clave: discondrosisplasia, condromatosis interna, encondromatosis o condromatosis múltiple familiar.
ABSTRAT
Multiple family enchondromatosis was described by Virchow in 1910, is presented with a pattern of autosomal dominant inheritance. The enchondromatosis comprise a heterogeneous group of congenital syndromes characterized by the presence of multiple enchondromas associated with musculoskeletal deformities secondary to shortening of the limbs. A family with four members affected, whose diagnosis was made by the clinical method through physical examination and imaging was presented. It is important to made an early diagnosis of this disease because of the possibility of a secondary prevention in patients as well as to provide appropriate genetic counseling to the family.
Keywords: discondrosisplasia, internal chondromatosis, enchondromatosis or family multiple chondromatosis.
INTRODUCCIÓN
]]> La osteocondromatis múltiple (OM) o encondromatosis múltiple familiar, es un trastorno hereditario. Esta patología fue descrita por Virchow, pero fue Ehrenfried quien publicó 12 casos y revisó la literatura en 1915 y 1917, y propuso el término “condrodisplasia deformante hereditaria” 1-3. En la literatura se describen las características de esta patología, como el ensanchamiento y deformación de la región metafisaria en comparación con la diafisaria, debido a una falla de la remodelación o trabeculación de esta región 2-5. Los sitios más comunes comprometidos son las regiones metafisaria alrededor de la rodilla, cadera y hombro.La deformidad más común es una combinación de disminución de la longitud del cúbito y arqueamiento de uno o de ambos huesos del antebrazo, inclinación cubital aumentada de la epífisis distal del radio (ángulo articular radial), desviación cubital de la mano y luxación proximal de la cabeza radial3. Las malformaciones músculo esqueléticas más frecuentes son: el acortamiento del cúbito con arqueamiento del radio (39-60%), crecimiento asimétrico de las extremidades (10-50%), baja estatura (37-44%), angulación en varo o valgo de la rodilla (8-33%) y deformidad de tobillo (2-54%)3-5.
Su cuadro clínico se caracteriza por el desarrollo de dos o más encondromas en las epífisis de los huesos largos, principalmente en la articulación femorotibial. Se presenta durante la primera década de la vida, con una edad media de diagnóstico a los tres años de edad. A diferencia de otras encondromatosis, una vez que se cierran los cartílagos de crecimiento, se detiene la formación de nuevos encondromas. La asimetría se presenta en un 70% de los pacientes, con problema ortopédico importante 5-7.
El diagnóstico de las encondromatosis se realiza bajo la sospecha clínica y se apoya en estudios de imagen. No existen pruebas específicas para su diagnóstico. Los pacientes en la infancia presentan deformidades esqueléticas, secundarias al desarrollo de los encondromas o la presencia de fracturas de forma patológica. La radiografía simple es el estudio inicial en la evaluación de los encondromas que generalmente se encuentran localizados en la diáfisis de los huesos largos y se observan como lesiones ovaladas, lobuladas y bien delimitadas 5-9.
El aspecto radiológico típico muestra zonas de osteólisis procedentes del cartílago de crecimiento, que se extienden hacia la diáfisis. Se pueden observar calcificaciones en copos de avena en un 50% de los casos6. Histológicamente la condromatosis múltiple se diferencia del condroma solitario por presentar un tejido más rico en células, con núcleos más grandes. El pronóstico dependerá de las complicaciones ortopédicas (asimetría de extremidades, deformidades, fracturas patológicas) y de la malignización de las lesiones 9.
Se han identificado dos genes supresores tumorales, involucrados en esta enfermedad: EXT1 y EXT2, localizados en los cromosomas 8q24 y 11p11-p12, respectivamente. Actualmente se sugiere la existencia de un tercer gen (EXT3) en el cromosoma 19p, sin embargo aún no ha sido identificado 10-14.
El crecimiento rápido de la lesión o el aumento de dolor aconsejan la evaluación y tratamiento precoz. El método de elección es entonces la resección tumoral asociada o no a un injerto u osteosíntesis. Este estudio se realiza a una familia con varios afectados que han sido intervenidos quirúrgicamente en varias ocasiones.
PRESENTACIÓN DE LOS CASOS
Se presenta una familia con tres generaciones donde el primer afectado (fallecido) corresponde al número I-1, no se pudo estudiar, solo se cuenta con fotos de las deformidades. No existen radiografías de ninguno de estas personas, a causa de que todas están muy dañadas y no ofrecieron información para presentar. El único descendiente el II-1 que presenta deformidades en los miembros superiores producto a los encondromas, esta paciente tuvo dos descendientes, una hembra y un varón igualmente afectados en la tercera generación. En todas las generaciones aparecen individuos afectados, con transmisión vertical, tanto hembras como varones, esto confirma la forma de transmisión autosómica dominante como se aprecia en la ( fig. 1).
]]>Descripción cada uno de los pacientes:
Pasiente 1
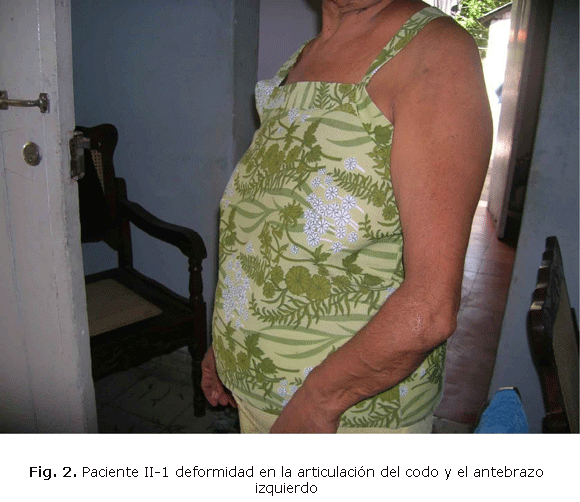
Femenina de 72 años, que corresponde en el árbol genealógico (fig.1) al paciente II-1, desde los 14 años presentó deformidad en la muñeca izquierda, en la derecha siempre fue menor. Los estudios imagenológicos determinaron que la deformidad era por encondromas, como se puede evaluar (fig.2). En la muñeca izquierda los encondromas continuaron su crecimiento produciendo impotencia funcional, operarse en varias ocasiones par mejorar la movilidad del miembro.
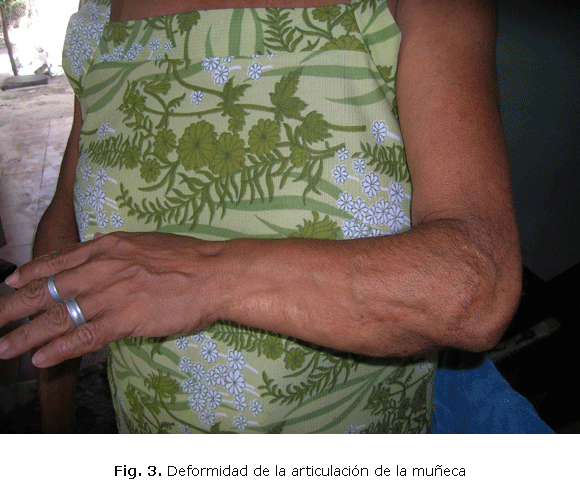
La deformidad de la articulación de la muñeca de la misma mano fue motivo de intervención quirúrgica en tres oportunidades por impotencia funcional (fig.3).
Pasiente 2
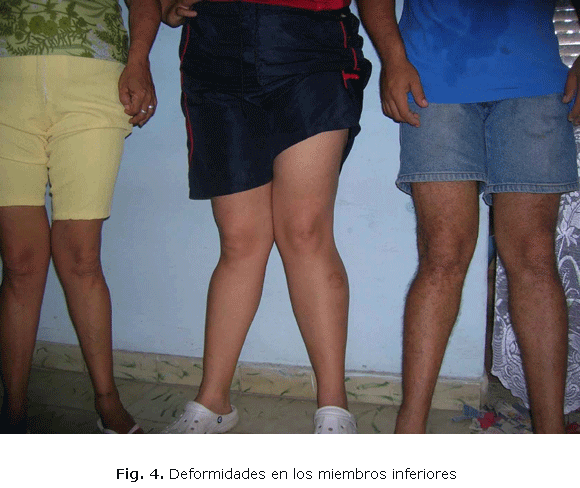
]]> Paciente femenina de 47 años de edad, que desde los tres años asistió a consulta de ortopedia por dolor y deformidad de ambas articulaciones de la rodilla; esto fue causa de caídas frecuentes. La deformidad continuó progresando como se observa en la (fig. 4), correspondiendo al sujeto III-1, de izquierda a derecha el primero corresponde al paciente II-1, la intermedia su hija III-1 y el último su hijo III-2.Pasiente 3
Paciente masculino de 42 años, que desde los cuatro años se atendió en consulta de ortopedia por caídas frecuentes y dolores articulares, las deformidades se presentaron alrededor de los 10 años, con crecimiento asimétrico de las extremidades inferiores, esto le impidió continuar estudios en la escuela becado donde le correspondía, operándose de la rodilla más afectada a los 16 años.
DISCUSIÓN
Esta enfermedad genética se describe con un patrón de herencia autosómica dominante 1-8 como lo presentado en esta familia. Las deformidades de antebrazo en la osteocondromatosis múltiple son frecuentes, siendo el cúbito el más afectado en su segmento distal, lo cual produce discrepancia de longitud del cúbito, traslación cubital del carpo y luxación o subluxación de la cabeza del radio 8, 9, como lo observado en uno de estos pacientes igualmente descrito por otros autores 10, 11.
Funcionalmente, estas deformidades se manifiestan con una pronosupinación limitada, la cual se incrementa cuando la cabeza del radio se luxa, lo que en un principio puede producir dolor y se puede observar prominencia de la cabeza del radio a nivel del codo. Un común denominador en todas estas situaciones es el arqueamiento del radio con discrepancia de longitud del cúbito. El 20% de los pacientes tienen luxación carpal, 19% un ángulo articular radial menor 30°, y 14% una luxación proximal de radio, como se describe en la paciente II-1.
Las lesiones raramente se advierten antes del primer año de edad, y es frecuente no precisarlas hasta el segundo o tercer año de vida. En general, las lesiones se vuelven más evidentes cuando crecen los niños, como ocurrió en los tres pacientes presentados, esto también lo refieren otros autores 5-9.
Por lo general se representa que una vez que termina el crecimiento, las lesiones no aumentan de tamaño, fenómeno que no ocurrió así en los pacientes descritos. Sin tratamiento durante el período de crecimiento, la discrepancia de longitud del cúbito progresa, el arqueamiento y el ángulo articular radial aumentan, la luxación carpal se incrementa, y la subluxación o luxación proximal radial es progresiva 6-10.
]]> El diagnóstico de las encondromatosis se establece por la sospecha clínica y los estudios imagenológicos, donde se aprecian los encondromas y la edad de inicio de aparición de estos y la evolución permite llegar al diagnóstico clínico definitivo. Resulta difícil establecer un diagnóstico diferencial cuando comienzan los primeros síntomas y las deformidades, pero después de los estudios y los antecedentes de familiares orientan hacia la encondromatosis múltiple familiar.La determinación genética de los genes EXT1 y EXT2 mediante microarreglos es el estudio más específico para el diagnóstico de la encondromatosis múltiple familiar 11-14, sin embargo, no en todos los hospitales se puede realizar esta práctica y, específicamente en Cuba, no se cuenta con este estudio. Es una limitación que tiene esta investigación para poder caracterizar molecularmente a estas familias y personas afectadas.
REFERENCIAS BIBLIOGRÁFICAS
1. Rozeman LB, Sangiorgi L, Briare de Brujin IH, Mainil Varlet P, Bertoni F, Cleton Jansen AM, et al. Enchondromatosis (Ollier Disease, Maffucci Syndrome) is not caused by the PTHR1 mutation p.R150C. Hum Mutat. 2004[citado 12 jun 2013]; 24(6):466-73. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/15523647
2. Ahmed SK, Lee WC, Irving RM, Walsh AR. Is Ollier’s disease an understaging of Maffucci’s syndrome? J Laryngol Otol. 1999[citado 10 Oct2013];113(9):861-4. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/10664698
3. Ramirez J, Padilla A, Romero A, Lavín A, Medina J, Dubón E, et al. Síndrome de Maffucci: Informe de dos pacientes y revisión de la literatura. Cir Ciruj. 2005; [citado 15 Ene 2014] 73: 217-21. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/16091163.
4. Desai S, Kubeyinje EP, Belagavi CS, Desai S. Maffucci’s syndrome. Ann Saudi Med 2007; [citado 18 Mar 2014] 17(4): 451-3. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/17353600.
5.Ryabykh SO, Gubin AV, Prudnikova CO, Kobyzev CA. Treatment of combined spinal deformity in patient with ollier disease and abnormal vertebrae. Global Spine J. 2013; [citado 11 Feb 2014]3 (2):109-14. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/24436859.
6. Fisher TJ, Williams N, Morris L, Cundy PJ. Metachondromatosis: more than just multiple osteochondroma. J Child Orthop. 2013; [citado 11Feb2014]7(6):455-64. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/24432109
7. McDermott AL, Dutt S, Chavda SV. Maffucci’s syndrome: clinical and radiological features of a rare condition. J Laryngol Otol. 2010; [citado 5 Mar 2014]115: 845-7. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/11668006
8. Dini LI, Isolan GR, Saraiva GA, Dini SA, Gallo P. Maffucci’s syndrome complicated by intracranial chondrosarcoma: two new illustrative cases. Arq Neuropsiquiatr. 2007; [citado 11 Feb 2014] 65(3): 816-21. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/17952287
9. Sutera R, Contiguglia A, Iovane A, Midiri M. A rare case of Enchondromatosis of the knees and hands with involvement of Hoffa's fat pad and peri-articular soft-tissues. J Radiol Case Rep. 2013 Jun 1 [citado 6 Ene 2014] 7(6):22-30. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/24421940
10. Gabos PG, Bowen JR. Epiphyseal-metaphyseal enchondromatosis. A new clinical entity. J Bone Joint Surg Am. 1998; [citado 11 Feb 2014] 80(6): 782-92. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/9655096
11. Tran Mau-Them F, Boualam A, Barat-Houari M, Jeandel C, Cottalorda J, Cormier-Daire V, et al Dysspondyloenchondromatosis without COL2A1 mutation: Possible genetic heterogeneity. Am J Med Genet A. 2014 [citado 23 May 2014] 164(3):769-73. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/ 24357493.
12. Nishio J, Kuwabara Y, Nabeshima S, Iwasaki H, Naito M. PET-positive polyostotic fibrous dysplasia mimicking Ollier disease. In Vivo. 2013; [citado 11 Feb 2014]27(6):821-6. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/24292588.
]]>13. Le BB, Nguyen BD. Ollier Disease With Digital Enchondromatosis: Anatomic and Functional Imaging. Clin Nucl Med. 2013 [citado 18 Mar 2014]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/24152634.
14. Hernández Rebeca G, Andina Martínez D, Pozo Román JA, Muñoz Calvo MT, Argente J. Granulosa cell tumour in a patient with multiple enchondromatosis. An Pediatr (Barc). 2014; [citado 11 Feb 2014] 80(3):86-7. Disponible en: http://linkinghub.elsevier.com/retrieve/pii/S1695403313003342
Recibido: 29 de octubre de 2013
Aprobado: 2 de diciembre 2013
Dra. Ana Iris Heres Zalazar. Centro Municipal de Genética Cacocum. Holguín. Cuba.
Correo electrónico: anairis@cacocum.hlg.sld.cu