
Caracterización clínica del síndrome de Usher. Provincia Holguín
Usher syndrome clinical characterization. Holguin province
Dra. Elayne Esther Santana HernándezI, Dr.C Paulina Araceli Lantigua CruzII.
I Centro Provincial de Genética Médica.Holguín.Cuba.
II Instituto de Ciencias Médicas de La Habana.Centro Nacional de Genética Médica.Cuba. ]]>
RESUMEN
Fundamento: El síndrome de Usher es una enfermedad determinada genéticamente, con una gran heterogeneidad clínica y genética; está caracterizada por hipoacusia neurosensorial de moderada a severa, retinosis pigmentaria progresiva y puede acompañarse de alteración vestibular. Por la alta prevalencia de esta enfermedad en la provincia de Holguín, se considera necesario este estudio.
Objetivo: Caracterizar clínicamente todos los enfermos con diagnóstico clínico de síndrome de Usher en la provincia Holguín, en el período de enero del 2009 a enero del 2016.
Metodología: Se realizó un estudio descriptivo, retrospectivo, tipo serie de casos, a los 53 pacientes con diagnóstico clínico de síndrome de Usher en la provincia Holguín. La muestra estuvo formada por los 53 enfermos residentes en la provincia. Se revisaron los registros del Centro Provincial de Retinosis Pigmentaria y las historias clínicas de estos pacientes; se recogieron los datos de interés en un instrumento que se confeccionó para ello. Las variables estudiadas fueron el sexo, la edad, edad del diagnóstico de la hipoacusia y severidad, edad del diagnóstico de la retinosis pigmentaria y los resultados de las pruebas audiológicas, lo que permitió conocer la función vestibular.
Resultados: Se caracterizó clínicamente el 100 % de los enfermos estudiados. Predominó el sexo masculino (60,37 %). El 80 % presentó la retinosis pigmentaria en la primera infancia y la hipoacusia congénita profunda en 67,92 %. Las pruebas vestibulares demostraron que el 71,70 % presenta síndrome de Usher tipo II y el 28,30 % tiene el tipo I.
Conclusiones: Predominó el sexo masculino, la hipoacusia precedió a la alteración visual. Se logró caracterizar clínicamente a estos afectados. Prevaleció el síndrome de Usher tipo II.
Palabras clave: Síndrome de Usher genética; retinitis pigmentosa; pérdida auditiva congénito; baja visión congénito; trastornos sordoceguera.
]]> DeCS: SÍNDROMES DE USHER/genética; RETINITIS PIGMENTOSA/genética; PÉRDIDA AUDITIVA/congénito; PÉRDIDA AUDITIVA SENSORINEURAL/congénito; BAJA VISIÓN/congénito; TRASTORNOS SORDOCEGUERA/genética.ABSTRACT
Background: Usher syndrome is a genetically determined disease with great clinical and genetic heterogeneity. This disease is characterized by sensorineural hearing loss of moderate to severe, progressive pigmentosa retinitis and may be accompanied by vestibular alteration. At the high prevalence of this disease in the province of Holguin, this study is considered necessary.
Objective: To characterize all patients clinically with clinical diagnosis of Usher syndrome in Holguin province, in the period from January 2009 to January 2016.
Methodology: A series types of retrospective cases, descriptive study with 53 patients with clinical diagnosis of Usher syndrome in Holguin province was conducted. The sample consisted of 53 patients residing in the province. Provincial records Pigmentosa Retinitis Pigmentosa Center and the medical records of these patients were reviewed, the data of interest are collected in an instrument that was drawn up for these. The variables studied were sex, age, age at diagnosis of hearing loss and severity, age of diagnosis of pigmentosa retinitis and the results of the audiological tests, allowing knowing the vestibular function.
Results: It was possible to clinically characterize 100 % of the patients studied, predominantly male in a 60.37 %. 80 % had pigmentosa retinitis in early childhood and profound congenital hearing loss in 67.92 %. Vestibular tests showed that 71. 70 % have Usher syndrome type II and 28.30 % have the type I.
Conclusions: mainly males, hearing loss preceded visual impairment. It was possible to clinically characterize those affected. It prevailed Usher syndrome type II.
Keywords: Usher syndrome/genetics; visual and hearing impairment and deafness retinitis pigmentosa; hearing loss/congenital; hearing loss sensorineural/congenital; vision low/congenital; deaf-blind disorders/genetics.
MeSH: USHER SYNDROMES/genetics; RETINITIS PIGMENTOSA/genetics; HEARING LOSS/congenital; HEARING LOSS SENSORINEURAL/congenital; VISION LOW/congenital; DEAF-BLIND DISORDERS/genetics.
]]>
INTRODUCCIÓN
El síndrome de Usher (USH), es una enfermedad genética de herencia autosómico recesiva, caracterizada por hipoacusia neurosensorial congénita asociada a retinosis pigmentaria progresiva y en ocasiones afectación de la función vestibular 1-2.
La prevalencia de la enfermedad en la población general varía desde 3,5 a 6,2 afectados por cada 100.000 habitantes, aunque ésta varía dependiendo de los países o regiones concretas en los que existen estudios epidemiológicos 3-5. Este síndrome es la causa más frecuente de sordo-ceguera en el mundo y corresponde 9,6 % de la población congénitamente sorda y al 18 % de toda la población con Retinosis Pigmentaria6-8.
Su clasificación clínica se basa en el grado de hipoacusia, edad de inicio de la retinosis pigmentaria (RP) y la posible afectación al sistema vestibular.9-11
Esta presenta una significativa heterogeneidad clínica y genética, ya que se conocen cuatro formas clínicas que a su vez se subdividen en subtipos genéticos, hasta el momento se han identificado 12 loci asociado: 7 del síndrome de Usher de fenotipo 1 (USH1B-H), 3 del síndrome de Usher de fenotipo 2(USH2A, 2C y 2D), 2 del síndrome de Usher de fenotipo 3(USH3A y 3B).
En la provincia de Holguín se encuentras varias familias afectadas, con 53 enfermos sin que existan estudios precedentes que permita clasificar clínicamente a estos enfermos. Estos pacientes enfermos se ven limitados escolarmente, pocos llegan a la educación superior, tienen una inadecuada ubicación laboral, carecen de ocupación de su tiempo libre y sufren por trastornos psicológicos. Por constituir esta enfermedad una problemática de salud en esta región, se considera necesario realizar un estudio que permita caracterizar a los afectados.
MATERIAL Y MÉTODO
Se realizó un estudio descriptivo, retrospectivo tipo serie de casos, a los 53 pacientes con diagnóstico clínico de síndrome de Usher en la provincia Holguín, en el período de enero del 2009 a enero del 2016.
]]> La muestra estuvo formada por los 53 enfermos residentes en la provincia Holguín. Se revisaron los registros del Centro Provincial de retinosis pigmentaria y las historias clínicas de estos pacientes. Se recolectaron datos clínicos de interés para desarrollar esta investigación, a través de las historias clínicas de estos enfermos. Las variables analizadas fueron: la edad (recogida por carnet de identidad y agrupada cada 9 años), el sexo masculino (M) y el femenino(F), edad inicio de la retinosis pigmentosa, considerado precoz antes de los 10 años y juvenil antes de los 20 años, se considera tardía después de los 30, pero al no tener algún afectado en este estadio no se incluyó en la tabla, y los grados de severidad de la alteración visual en: grado I , grado II, grado III y grado IV, este último se considera en amaurosis total o ceguera. La edad al diagnóstico de la hipoacusia y grado de severidad, considerándose congénita la hipoacusia al nacimiento, primera infancia antes de los 10 años, adolescencia después de los 11 años, adultez después de los 20 años y tardía después de los 30 años no diagnosticándose enfermo alguno a esta edad.Se realizó una prueba calórica para conocer la función vestibular, se consideró normal cuando el paciente tiene respuesta (nistagmos) y alterada cuando no tiene respuesta; esta prueba audiológica permitió clasificar a estos pacientes con síndrome de Usher tipo I, tipo II y tipo III.
Se utilizaron estadígrafos de medianas y porcientos para la confección de las tablas.
RESULTADOS
Casi la mitad de los pacientes tiene una edad comprendida entre los 40 y 59 años un total de 27 afectados (50,93 %), asimismo predominó el sexo masculino con 32 enfermos (60,37 %), como se aprecia en la tabla 1.
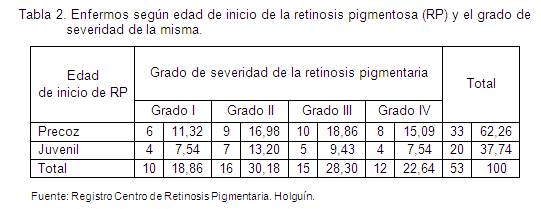
En la tabla 2 se relacionan la edad de inicio de la retinosis pigmentaria y el grado de severidad, donde 31 pacientes se encuentran en grado de moderado a severidad en estadio II y III, identificándose una afectación visual avanzada como se espera en estos afectados.
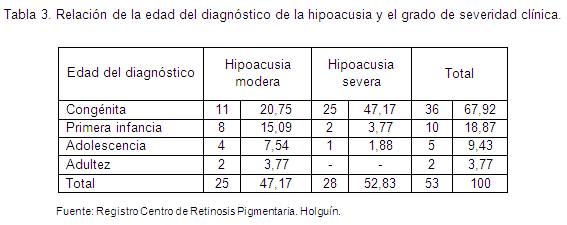
Resulta imprescindible en esta enfermedad conocer el resultado de los estudios de audiología, donde la edad del diagnóstico de la hipoacusia y el grado de severidad es un dato de interés, por esto se relacionan en la tabla 3. Se encontraron 36 pacientes con hipoacusia congénita de moderada a severa (67, 92 %).
Respuesta vestibular ante estimulación calórica:
DISCUSIÓN
El síndrome de Usher es una enfermedad hereditaria autosómica recesiva, lo que significa que los padres son personas sanas portadoras de mutaciones para esta afección, que cuando dos portadores con esta condición contraen matrimonio pudieran tener la probabilidad de que nazcan hijos enfermos en un 25 %, una probabilidad alta de que aparezcan individuos afectados, aún sin conocerse. 1-3
Se ha descrito que el USH muestra una amplia heterogeneidad genética dada por la cantidad de mutaciones que se han secuenciado en esta enfermedad e izoformas que ya se consideran mutaciones nuevas 1-3. Sin embargo para realizar un estudio molecular es necesario tener un estudio anterior donde estén caracterizados clínicamente los afectados 4-6.
Los trastornos auditivos anteceden a los visuales, como lo descrito por otros autores 6. Se logró confirmar la hipoacusia neurosensorial bilateral moderada a profunda en la totalidad de los enfermos, resultado que coincide con otros estudios 7-9. Se diagnosticó la mayoría como hipoacusia congénita severa y otro grupo en la primera infancia siendo este el primer signo clínico, como lo que se ha planteado en estos afectados.10-12
A través de las pruebas vestibulares se logra la clasificación clínica de estos pacientes, en las que se encontró respuesta normal en 38 pacientes, se clasificó este grupo como síndrome de Usher tipo II; y en 15 enfermos con estudio alterado, síndrome de Usher tipo I, lo que coincide con lo descrito en la literatura 9-11.
]]> El comienzo de las manifestaciones de retinosis pigmentaria fue precoz en 33 pacientes (62,26 %), el grado de severidad prevaleció en el estadio II y III; solo 12 afectados están en estadio IV de amaurosis o ceguera total. La pérdida del campo visual se comporta con gran variabilidad intrafamiliar, es decir se observó que en una misma familia varios enfermos de un mismo tipo clínico presentan diferentes estadios de pérdida visual por retinosis pigmentaria, como describen otros autores 13,14.En otras investigaciones realizadas han podido caracterizar molecularmente sus afectados han encontrado nuevas mutaciones y cambios conformacionales en otros genes asociados a retinosis pigmentaria que pudieran actuar como genes modificadores de la expresión fenotípica en la pedida visual, esto pudiera explicar en parte la variabilidad encontrada 11,12.
En Cuba por la colonización por los españoles, existe la probabilidad de que se haya introducido una mutación ancestral, por lo que sería muy conveniente comenzar los estudios moleculares para buscar las mutaciones encontradas en investigaciones realizadas en esa población 14. Es probable que estos genes traídos por los europeos producto de la colonización se expandieran en las Américas y esta pudiera ser la causa de que la prevalencia del síndrome Usher tipo II sea alta en Latinoamérica 4,6,7.
Se considera necesario realizar estudios que permitan caracterizar clínicamente a estos enfermos para poder realizar en otro momento estudios moleculares.
CONCLUSIONES
Predominó el sexo masculino, la hipoacusia precedió a la alteración visual. Se logró caracterizar clínicamente a estos afectados. Prevaleció el síndrome de Usher tipo II.
REFERENCIAS BIBLIOGRÁFICAS
1. Sadeghi AM, Cohn ES, Kimberling WJ, Halvarsson G, Möller C. Expressivity of hearing loss in cases with Usher syndrome type IIA. Int J Audiol. 2013 Dec; [citado 11 Feb 2014] 52(12):832-7. Disponible en:http://www.ncbi.nlm.nih.gov/pubmed/24160897.
2. Boo SH, Song MJ, Kim HJ, Cho YS, Chu H, Ko MH, Chung WH, Kim JW, Hong SH. A Novel Frameshift Mutation of the USH2A Gene in a Korean Patient with Usher Syndrome Type II. Clin Exp Otorhinolaryngol. 2013 Mar; [citado 3 Ene 2014]6(1):41-4 Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/23526569
3. Praharaj SK, Acharya M, Sarvanan A, Kongasseri S, Behere RV, Sharma PS. 10. Mania associated with Usher syndrome type II. Turk Psikiyatri Derg. 2012 Fall; [citado 8 Ene 2014] 23(3):219-21. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/22949292
4. López G, Gelvez NY, Tamayo M. Mutational frequencies in usherin(USH2A gene) in 26 Colombian individuals with Usher syndrome type II. Biomedica. 2011 Mar; [citado 12 Mar 2012]31(1):82-90. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/22159486
5. Wang L, Zou J, Shen Z, Song E, Yang J. Whirlin interacts with espin and modulates its actin-regulatory function: an insight into the mechanism of Usher syndrome type II. Hum Mol Genet. 2012 Feb 1; [citado 12 Mar 2013]21(3):692-710. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/22048959
6. Garcia-Garcia G, Aparisi MJ, Jaijo T, Rodrigo R, Leon AM, Avila-Fernandez A, Blanco-Kelly F, Bernal S, Navarro R, Diaz-Llopis M, Baiget M, Ayuso C, Millan JM, Aller E. Mutational screening of the USH2A gene in Spanish USH patients reveals 23 novel pathogenic mutations. Orphanet J Rare Dis. 2011 Oct 17; [citado 12 Mar 2012]6:65. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/22004887
7. Tamayo ML, Lopez G, Gelvez N, Medina D, Kimberling WJ, Rodríguez V, Tamayo GE, Bernal JE. Genetic counseling in Usher syndrome: linkage and mutational analysis of 10 Colombian families. Genet Couns. 2008; [citado 12 Mar 2012]19(1):15-27. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/18564497
8. Lenassi E, Saihan Z, Bitner-Glindzicz M, Webster AR. The effect of the common c.2299delG mutation in USH2A on RNA splicing. Exp Eye Res. 2014 May; [citado 17 Abr 2013]122:9-12. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/24607488.
9. Aller E, Larrieu L, Jaijo T, Baux D, Espinós C, González-Candelas F, Nájera C, Palau F, Claustres M, Roux AF, Millán JM. The USH2A c.2299delG mutation: dating its common origin in a Southern European population. Eur J Hum Genet. 2010 Jul; [citado 8 Ene 2014]18(7):788-93. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/20145675.
10. Yan D, Ouyang X, Patterson DM, Du LL, Jacobson SG, Liu XZ. Mutation analysis in the long isoform of USH2A in American patients with Usher Syndrome type II. J Hum Genet. 2009 Dec; [citado 8 Ene 2014]54(12):732-8. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/19881469.
11. Dreyer B, Tranebjaerg L, Rosenberg T, Weston MD, Kimberling WJ, Nilssen O. 10. Identification of novel USH2A mutations: implications for the structure of USH2A protein. Eur J Hum Genet. 2000 Jul; [citado 8 Ene 2014]8(7):500-6. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/10909849.
12. Ouyang XM, Hejtmancik JF, Jacobson SG, Li AR, Du LL, Angeli S, Kaiser M, Balkany T, Liu XZ. Mutational spectrum in Usher syndrome type II. Clin Genet. 2004 Apr;65(4):288-93. Erratum in: Clin Genet. 2004 May; [citado 8 Ene 2014]65(5):433. Yam, D Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/15025721
13. Dreyer B, Tranebjaerg L, Brox V, Rosenberg T, Möller C, Beneyto M, Weston MD, Kimberling WJ, Cremers CW, Liu XZ, Nilssen O. A common ancestral origin of the frequent and widespread 2299delG USH2A mutation. Am J Hum Genet. 2001 Jul;69(1):228-34. Epub 2001 Jun 8. Erratum in: Am J Hum Genet 2001 Oct; [citado 8 Ene 2014]69(4):922. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/11402400.
14. Aller E, Nájera C, Millán JM, Oltra JS, Pérez-Garrigues H, Vilela C, Navea A, Beneyto M. Genetic analysis of 2299delG and C759F mutations (USH2A) in patients with visual and/or auditory impairments. Eur J Hum Genet. 2004 May; [citado 8 Ene 2014]12(5):407-10. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/14970843
Recibido: 2015-05-28 ]]> Aprobado: 2016-10-26
Dra.Elayne Esther Santana Hernández. Centro Provincial de Genética Médica. Holguín. Cuba.
]]>