Se reportan los árboles genealógicos de 10 probandos afectados con sorderas neurosensoriales no sindrómicas de aparición familiar. El análisis genético practicado permitió reconocer la clara segregación de un único gen de sordera en 7 familias (3 autosómicas recesivas, 2 autosómicas dominantes, 1 ligada al cromosoma X, 1 con herencia mitocondrial). En las 3 familias restantes resultó difícil el análisis y se propuso la herencia recesiva como la más probable, sobre la base, fundamentalmente, de las características de la pérdida auditiva (congénita, bilateral, severa o profunda). En general las sorderas autosómicas recesivas fueron las más frecuentes. Se corrobora que estos estudios suelen ser complicados por la gran heterogeneidad que pueden presentar a todos los niveles las sorderas neurosensoriales no sindrómicas.
Descriptores DeCS: SORDERA/genética.
Las sorderas hereditarias afectan aproximadamente a 1 de cada 2 000 nacidos y representan un serio problema de salud pública en todos los países.1-3
Las sorderas no sindrómicas son aquéllas en que la hipoacusia no se asocia con otras características clínicas y constituyen el 70 % de pacientes con sorderas hereditarias.1,4 Como se conoce, en las sorderas neurosensoriales la afectación de la transmisión del sonido se produce a nivel de cóclea o de nervio auditivo.5
El análisis genético en las familias en las cuales se ha demostrado la segregación de un solo gen, ha permitido clasificar las sorderas neurosensoriales no sindrómicas en autosómicas o ligadas al cromosoma X y éstas a su vez en recesivas o en dominantes.1,6
En este trabajo presentamos 10 "probandos" con formas heredadas de sorderas neurosensoriales no sindrómicas. Se analizan algunas características clínico-audiológicas y genéticas de ellas y se hacen consideraciones acerca de la utilidad actual de estos estudios.
MÉTODOS
La investigación se basó en los aspectos siguientes:
Examen físico general.
Confección del árbol genealógico.
Examen audiológico.
El examen físico se realizó por un genetista clínico, con la finalidad de conocer las alteraciones físicas o signos asociados con la hipoacusia. Se confeccionó el árbol genealógico a partir del "probando" afectado por sordera neurosensorial de presentación familiar sin otra afectación clínica acompañante. Los datos se obtuvieron al interrogatorio de 2 o más familiares. Se procedió al examen audiológico de todos los individuos (sanos y enfermos) posibles de cada familia. ]]>
El diagnóstico de la hipoacusia se efectuó por un especialista en ORL y se basó en las siguientes investigaciones según el caso:
Exploración general otorrinolaringológica.
Acumetría con pruebas de Rinne, Weber y Schwabach.
Estudio audiométrico.
Impedanciometría con registros del timpanograma y estudio del reflejo estapedial
Potenciales evocados auditivos de tallo cerebral.
RESULTADOS
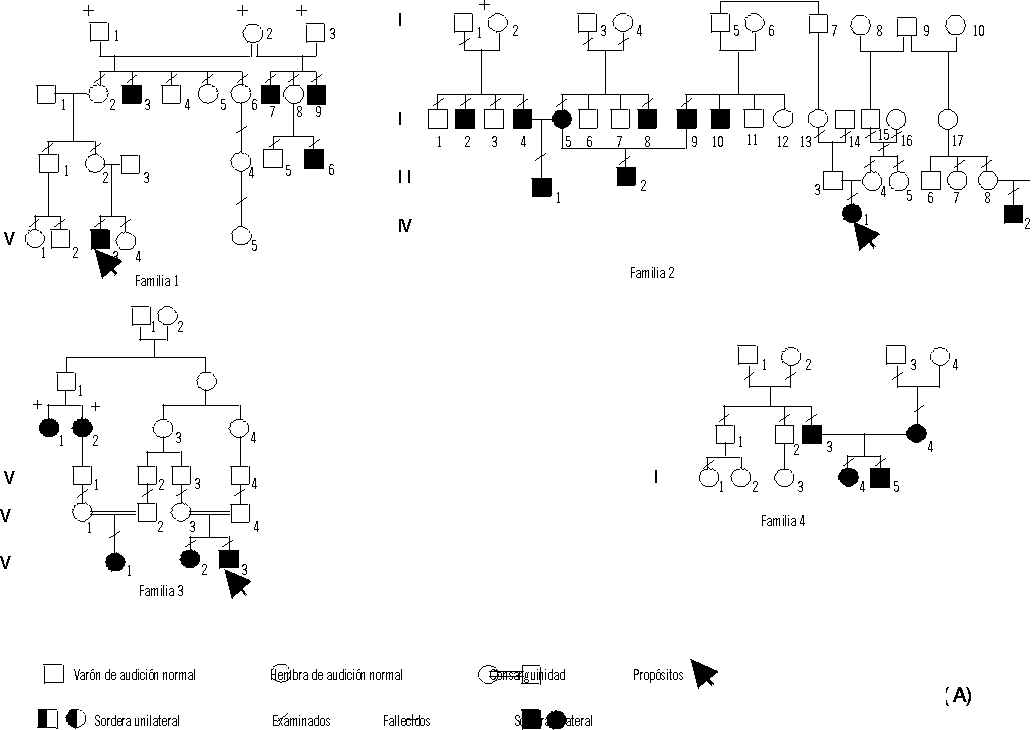
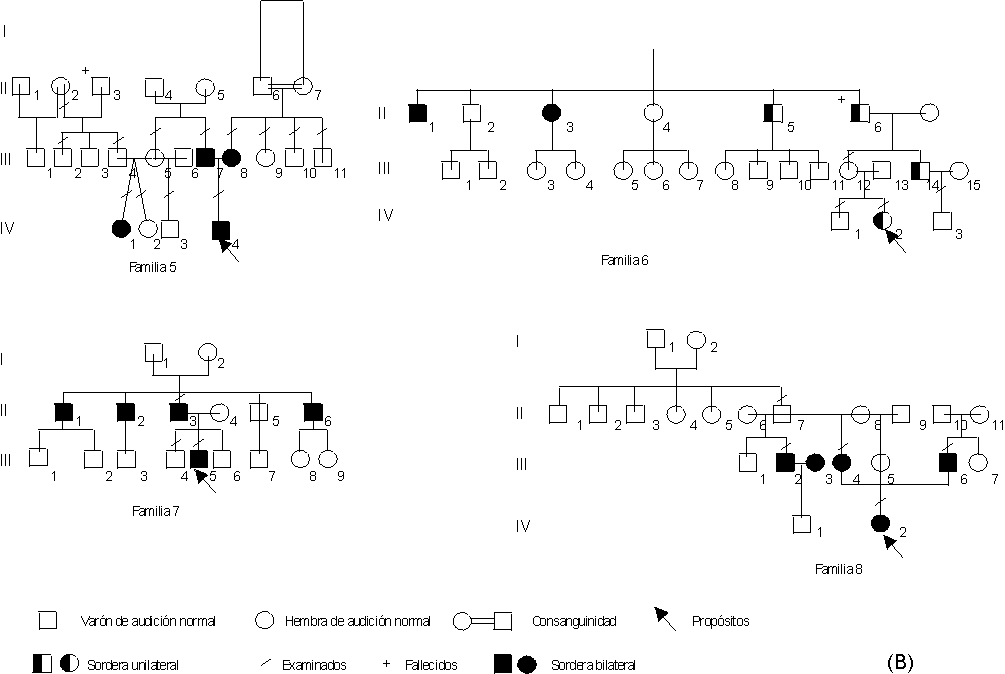
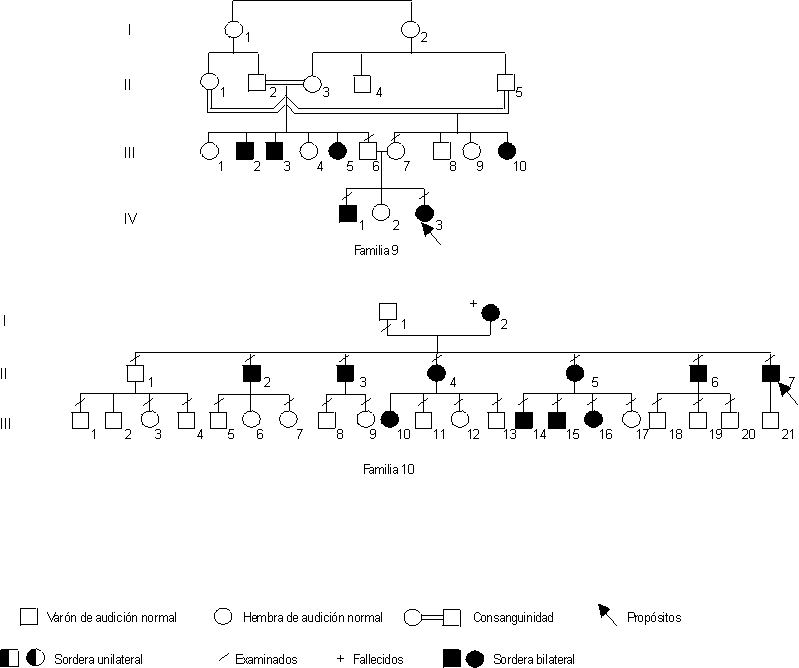
Éstos se expresan según los "probandos" y las 10 familias estudiadas (figs. A, B y C).
]]>
Fig. Arboles genealógicos de las familias.
Familia 1. Paciente IV-3. Niño de 7 años en el cual se sospechó la pérdida auditiva desde pocos meses de nacido. Ésta se caracterizó como neurosensorial profunda congénita y bilateral. Tres tíos y un primo por vía materna eran sordomudos. Se diagnosticó como sordera neurosensorial congénita ligada al cromosoma X.
Familia 2. Paciente IV-1. Niña de 1 año de edad que nació de un parto normal. A los 8 meses de nacida le notaron que no oía bien. Se le realizaron los estudios correspondientes y se le diagnosticó una pérdida auditiva neurosensorial bilateral profunda. Ambos padres, con audición normal, refirieron tener familiares sordomudos. El diagnóstico fue sordera neurosensorial congénita bilateral autosómica recesiva.
Familia 3. Paciente VI-3. Niño de 3 años de edad que se le diagnosticó hace un año sordera congénita neurosensorial bilateral profunda. Ambos padres son consanguíneos. La hermana y una prima presentan un cuadro similar. Se clasificó como sordera neurosensorial congénita autosómica recesiva.
Familia 4. Paciente III-5. Paciente de 8 meses de edad que concurre a consulta con sus padres, los cuales son sordomudos, para precisar su estado auditivo. Se le detecta pérdida auditiva bilateral neurosensorial profunda, al igual que sus padres y hermana mayor. No existen otros antecedentes familiares de la afección, a no ser los referidos. Se caracterizó como una posible sordera neurosensorial congénita autosómica recesiva.
Familia 5. Paciente IV-4. Niño de 11 años de edad nacido de embarazo y parto normales. Ambos padres y una prima paterna son sordomudos. En los 3 la sordera se había catalogado como de causa ambiental por el uso de medicamentos ototóxicos. Los abuelos maternos son consanguíneos y en él no se refirió el uso de medicamentos con toxicidad demostrada al aparato auditivo. Se propone descartar la herencia autosómica recesiva.
Familia 6. Paciente IV-2. Paciente de 9 años de edad en los que se niegan antecedentes de interés pre y posnatales que pudieran estar relacionados con la enfermedad en cuestión de dicha paciente. Consulta porque "no oye nada" por el oído derecho. Los estudios realizados destacan pérdida auditiva neurosensorial severa por ese oído. Existen miembros de la familia con pérdida auditiva neurosensorial profunda congénita bilateral y otros con hipoacusia del mismo tipo, pero unilateral. Se concluye como sordera neurosensorial autosómica dominante con expresividad variable y penetrancia reducida.
Familia 7. Paciente III-5. Es un niño que a los 5 años de edad sus padres le notaron que lateralizaba la cabeza para escuchar lo que le hablaban. Los estudios audiológicos detectaron una hipoacusia neurosensorial bilateral, con mayor afectación del oído izquierdo. Ahora tiene 12 años de edad y los chequeos periódicos han demostrado que la pérdida auditiva es progresiva. Su padre y 3 tíos paternos refieren pérdida auditiva bilateral a partir de la 2da. o 3ra. década de la vida. Aunque la hipoacusia sólo se ha manifestado en 2 generaciones, se considera del tipo neurosensorial progresiva autosómica dominante con penetrancia reducida.
Familia 8. Paciente IV-2. Niña de 4 meses de edad a la que se le realizan estudios de pesquisaje auditivo por mostrar antecedentes familiares de sordera. Se le detecta pérdida auditiva neurosensorial congénita bilateral profunda. La madre y el tío materno son sordomudos. El padre presenta una hipoacusia neurosensorial bilateral moderada sin antecedentes demostrados y ha desarrollado lenguaje; es el único afectado en su familia. Se propone la herencia autosómica recesiva que se deberá investigar posteriormente con otros estudios. ]]>
Familia 9. Paciente IV-3. Paciente de 10 años y del sexo femenino. Destaca sordera neurosensorial bilateral congénita, profunda en el oído derecho y severa en el izquierdo. Existen otros miembros de la familia afectados con consanguinidad demostrada. Se concluye como sordera neurosensorial congénita autosómica recesiva.
Familia 10. Paciente II-7. Adulto de 50 años de edad que comenzó con pérdida auditiva durante la segunda década de la vida. El déficit neurosensorial fue bilateral -aunque mayor del oído izquierdo- y progresivo. Actualmente la pérdida puede considerarse de severa a profunda que afecta a todas las frecuencias, en ambos oídos. Cinco de sus hermanos presentan también pérdida progresiva de la audición de comienzo en la primera o segunda décadas de la vida. Sólo las hembras afectadas han tenido descendencia con esta enfermedad. No se reporta consanguinidad parental. Se clasifica como sordera neurosensorial progresiva con patrón de herencia mitocondrial.
DISCUSIÓN
El análisis genético practicado en estas familias ha permitido reconocer a la herencia monogénica como causa de sorderas neurosensoriales en nuestro medio. En 5 familias fueron genes autosómicos y en 2, localizados en el cromosoma X y en la mitocondria respectivamente. Para las 3 fa-milias restantes, familias 4, 5 y 8 (fig.) propusimos la herencia autosómica recesiva, para ello nos basamos fun-damentalmente, en las características de la pérdida auditiva en los "probandos" y sus padres y por razones de frecuencia.1,6-8 Los matrimonios entre sordos pueden dar lugar a la presencia de 2 genes distintos de sordera en la misma familia -heterogeneidad genética alélica o no-, lo que dificulta extraordinariamente el análisis genético en éstas.7-9 Todas las sorderas recesivas que se han reportado con localización cromosómica conocida resultaron congénitas, o prelinguísticas y severas o profundas.6,9-12. Hasta el momento sólo se ha reportado un tipo de sordera neurosensorial prelinguística, no progresiva, que afecta a todas las frecuencias con patrón de herencia autosómica dominante, en la misma región donde se ha localizado el gen de la sordera recesiva NSDR1. Esto ha llevado a sugerir que mutaciones en el mismo gen pudieran dar lugar a sorderas de tipo dominante o recesivo.13 Muchas de estas familias de difícil interpretación genética hoy día y aquéllas de causa no precisada, pudieran ser caracterizadas en un futuro mediante estudios moleculares.10 En la familia 10 el patrón de herencia nos hizo sospechar desde un inicio la existencia de una mutación en el genoma mitocondrial,14,15 lo cual se confirmó molecularmente y será motivo de un trabajo ulterior.
Los estudios genéticos-moleculares en las sorderas neurosensoriales no sindrómicas han experimentado un considerable desarrollo en los últimos 5 años. La localización cromosómica de genes implicados en ellas reportan los resultados siguientes:9-20
Diez genes de sorderas autosómicas recesivas en: 2p, 3p, 7q, 9q, 10q, 11q, 13q, 14q, 17p y 21q.
Once genes de sordera autosómica dominante en: 1p, 1q, 4p, 5q, 6q, 7p, 11q, 13q, 14q, 15q y 19q.
Cuatro genes de sordera ligada al cromosoma X en: Xq21.1, Xq21.2, Xq22 y Xp22.
Como se ve, el reconocimiento de estas familias afectadas con sorderas de causa claramente genética constituye además, un material biológico de valor inapreciable para realizar estudios de ligamiento que confirmen las localizaciones ya reportadas10 o para la identificación de nuevos loci. Tales estudios permitirán en un futuro disponer de protocolos de diagnósticos y asesoramientos genéticos más refinados, que contribuyan a la prevención de las sorderas hereditarias, además de ampliar los conocimientos en su fisiopatología molecular y posible terapéutica.
Summary
The pedigress of 10 examiness affected with non syndrome neurosensorial deafness of familial appearance were erported. The genetical analysis made allowed to recognize the clear segregation of a sale gene of deafness in 7 families (3 recessive autosomal, 2 dominant autosomal, 1 linked to cromosome X, and 1 with mitochondrial heredity). In the other 3 hamilies the analysis was difficult and the recessive heredity was suggested as the most probable, based mainly on the characteristics of auditive loss (congenital, bilateral, severe or deep). In general, the recessive autosomal deafness was the most common. It was corroborated that these studies are usually complicated due to the great heterogeneity that the non syndromic neurosensorial deafness may present at all levels.
Subject headings: DEAFNESS/genetics.
REFERENCIAS BIBLIOGRÁFICAS
Reardon W. Genetic deafness. J Med Genet 1992;29:521-6.
Morton NE. Genetic epidemiology of hearing impairment. Ann N y Acad Sci 1991;630:16-31.
]]>
Xuezhong L, Lirong X, Silig Z, Ying X. Prevalence and aetiology of profound deafness in the general population of Sichuan, China. J Laryngol Otol 1993;107:990-3.
Bergstrom L, Hemennway W, Downs M. A high risk registry to find congenital deafness. Otolaryngol Clin North Am 1971;4:369-99.
Nadol Jb. Hearing loss. N Engl J Med 1993;1092-101.
Arnos KS, Israel J, Devlin L, Wilson MP. Genetic counseling for the deaf. Otolaryngol Clin North Am 1992;25:953-71.
Arnos KS, Cunningham M, Israel J, Marazita L. Innovative approach to genetics counseling services for the deaf population. Am J Med Genet 1992;44:345-51.
Moreno F, Castillo I del Villamar M. Las sorderas hereditarias. Nuevo objetivo de la genética molecular. FIAPAS 1995;47:37-9.
Petit C. Gene responsible for human hereditary deafness: symphony of a thousand. Nature Gen 1996;14:385-91.
Guilford P, Ben Arab S, Blanchard S, Levilliers J, Weisenbach J, Belkahia A, et al. A non syndromic form of neurosensory recessive deafness map to the pericentromeric region of chromosome 13q. Nature Gen 1994;6:24-8.
Friedman TB, Liang Y, Weber TL. A gene for congenital recessive deafness DFNB3 maps to the pericentromeric region of chromosome 17. Nature Gen 1995;9(1):86-91.
]]>
Chaib H, Lina-Grade G, Guilford P. A gene responsible for a dominant form of neurosensory non-syndromic deafness maps to the NSRD1, recessive deafness gene interval. Hum Mol Genet 1994:3(12):2219-22.
Jaber L, Shohat M, Bu X, Fishel-Ghodsian N, Yang H, Wang S, et al. Sensorineural deafness inherited as a tissue specific nitochondrial disorder. J Med Genet 1992;29:86-90.
Gold M, Rapin I. Non mendelian mitochondrial inheritance as a cause of progressive genetic neurosensorial hearing loss. Int J Pediatr Otorhinolayngol 1994;30:91-104.
Piussan C, Hanauer A, Dahl N, Mathieu M. X linked progressive mixed deafness: a new microdeletion that involves a more proximal region in Xq21. Am J Hum Genet 1995;56:224-30.
Lalwani AK, Brister R, Fex J, Grunfast K, Pikus A, Ploptis B, et al. A new non-syndromic X-linked sensorineural hearing impairment linked to Xp21.2. Am J Hum Genet 1994;55:685-94.
de Kok Y, Maarel SM Van der, Bitner-Glindzicz M, Huber I, Monaco A, Malcom S, et al. Association between X-linked mixed deafness mutations in the POU Domain gene POU3F4. Sciences. 1995;267:685-8.
Tyson J, Bellman S, Newton V, Simpson P, Malcom S, Pembrey M, et al. Mapping of DFN2 to Xq22. Human Mol Gen 1996;5:2055-60.
Castillo I del Villamar M, Sarduy M, Romero L, Herraiz C, Hernández F, et al. A vovel locus for non-syndromic sensorineural deafness (DFN6) maps to chromosome Xp22. Human Mol Gen 1996,5:1383-7.
Recibido: 6 de junio de 1996. Aprobado: 23 de octubre de 1997.
Dra. Ibis Menéndez. Hospital Pediátrico Docente "William Soler", Calle 100 y Perla, Boyeros, apartado 8019, Habana 8, Ciudad de La Habana, Cuba. ]]>
1Departamento de Genética. Hospital Pediátrico Docente "William Soler". 2 Departamento de Audiología. Hospital Pediátrico Docente "William Soler". 3 Hospital General Docente "Aleida Fernández Chardiet". ]]>199229521-6199163016-311993107990-319714369-9919931092-101199210199225953-71199244345-5119954737-9199614385-911994624-8199591186-9119943(12)(12)2219-2219922986-9019943091-104199556224-30199455685-941995267685-8199652055-60199651383-7