RESUMEN
Como enfermedades renales poliquísticas hereditarias se describen clásicamente la autosómica recesiva y la autosómica dominante, mal llamadas enfermedad poliquística de «tipo infantil» y de «tipo adulto», respectivamente, pues ambas pueden verse tanto en una como en otra edad. Los conceptos cambiantes en cuanto a la enfermedad autosómica recesiva, dados por los progresos en el tratamiento de los recién nacidos con la enfermedad, y la localización del gen, que por su mutación la produce, nos motivan hacer esta breve revisión con la finalidad de contribuir a la comprensión de la enfermedad por los estudiantes de medicina y el médico general básico.
Palabras clave: Enfermedad poliquística autosómica recesiva, fibrosis hepática congénita, quistes renales.
La enfermedad poliquística autosómica recesiva (EPAR) está asociada a una disgenesia biliar conocida como «fibrosis hepática congénita», cuyo gen (denominado PKHD1, que es la sigla formada de su nombre en inglés) está localizado en el cromosoma 6 (6p21).1-4 No se conoce exactamente su incidencia, pero algunos autores la han estimado entre 1 en 10 000 a 40 000 nacidos vivos,1,2 mientras otros señalan 1 por cada 20 000 nacimientos.5-7 Esta es una causa poco frecuente de insuficiencia renal en el niño,1 y se ha observado una variabilidad fenotípica considerable, incluso dentro de una misma familia, desde grandes riñones quísticos con muerte perinatal hasta fibrosis hepática congénita con afectación renal mínima.8
Como enfermedad recesiva típica, los heterocigotos no tienen síntomas; uno y otro sexo se afectan por igual y el riesgo de repetición es de 25 % en cada uno de los embarazos subsiguientes de las parejas en riesgo.2 Los pacientes que vivan largo tiempo hasta tener la posibilidad de ser padres no corren el riesgo de tener hijos con la enfermedad, si su pareja no tiene relación genética.9
Refieren Ong y Wheatley que en 1841 se describió por vez primera la degeneración quística de los riñones; en 1888, los riñones poliquísticos como un síndrome, y en 1899 se notó la naturaleza hereditaria en algunas familias.10
]]> La EPAR fue reconocida morfológicamente en 1902, pero sus características hísticas no se describieron hasta 1947.11 En 1964, Osathanondh y Potter clasificaron la EPAR como la enfermedad quística renal de tipo 1,12 y no fue hasta 1994 que se identificó el locus de la enfermedad.13En 1971, Blyth y Ockenden publican una clasificación basada en la edad y forma de presentación, las anomalías asociadas y el porcentaje de túbulos afectados y pronóstico de esta.14 Dicha clasificación divide la enfermedad en cuatro categorías: perinatal, neonatal, infantil y juvenil.
Esta clasificación, que sólo contemplaba los niños, con los avances en el estudio ultrasonográfico (que en muchos casos permite un diagnóstico precoz), la mejoría de los cuidados intensivos perinatales que han modificado la evolución a corto plazo, y el hallazgo de la mutación genética que produce la enfermedad, ha quedado obsoleta por no corresponderse con la observación actual, según la mayoría de los entendidos,1,4,6-8,15,16 pero aún puede ayudar en el caso de los recién nacidos.
La enfermedad responde a mutaciones en el gen PKHD1 en el cromosoma 6p21, que codifica la fibrocistina/poliductina y, aunque la mayoría de los casos se manifiestan en las etapas perinatal y neonatal, el espectro clínico de los pacientes que sobreviven estos períodos de la vida es mucho más variable de lo que se pensaba hace pocos años.4
La severidad fenotípica entre las mujeres sugirió que las formas más graves de la enfermedad podían deberse a mutaciones en un segundo gen, pero en un estudio de 22 familias con formas perinatales graves, las mutaciones sólo se encontraron en el mismo locus del cromosoma 6 entre la banda p21 y su centrómero.17
Histológicamente hay una marcada ectasia de los tubos colectores, los cuales están dispuestos en posición radial desde el cáliz a la cápsula, limitados por un epitelio plano o cuboidal. Cuando avanza la edad se pierde esta uniformidad, porque los quistes más grandes comprimen los cálices. Pueden encontrarse quistes glomerulares. Hay pequeñas áreas de parénquima normal en la corteza sin evidencia de displasia, aunque se han reportado células musculares lisas alrededor de los quistes. Durante la enfermedad aparece fibrosis intersticial, atrofia tubular y esclerosis glomerular, pero se desconoce el mecanismo de su producción.1
Los quistes de la EPAR se originan del túbulo proximal en el primer trimestre del embarazo, pero al nacimiento están localizados exclusivamente en los tubos colectores y mantienen su continuidad con la nefrona.18,19
Aunque clásicamente los dos riñones están aumentados de tamaño, se han reportado casos de asimetría renal.20
El espectro clínico varía desde el mortinato o deceso neonatal hasta la supervivencia en la vida adulta.21,22 Los casos más graves pueden manifestarse por oligohidramnios antes del nacimiento.1 Muchos pacientes presentarán masas renales palpables, dificultad respiratoria y, por el gran tamaño de los riñones con compresión diafragmática y del estómago, pueden mostrar dificultad para la alimentación.23 Como manifestación más frecuente en los niños que rebasan el período neonatal se halla la hipertensión arterial, que se presentará entre 55 % y 75 % de los casos,4,6,7,22 y de la cual se han destacado su magnitud y su difícil manejo.24
La hipertensión portal es el problema principal que se asocia a la fibrosis hepática congénita y causa hiperesplenismo y várices esofágicas. Un grupo de pacientes también muestra dilatación quística de los conductos biliares (síndrome de Caroli), con riesgo de colangitis recurrente.25,26
Los casos más graves pueden sospecharse prenatalmente por oligohidramnios y vejiga vacía. Las formas más ligeras pueden no detectarse mediante el ultrasonido materno-fetal, pero en este estudio puede encontrarse aumento de volumen e hiperecogenicidad renal, hallazgo que aparecerá entre las 20 y 34 semanas de gestación, por lo que algunos casos no son diagnosticados. El diagnóstico prenatal ha aumentado considerablemente, y Gagnadoux y Broyer reportan 20 % (5/25) entre 1962 y 1979 y 69 % (11/16) entre 1980 y 1990.1
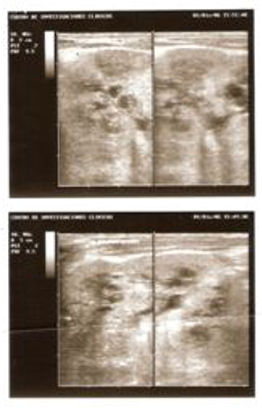
Los resultados del ultrasonido materno-fetal pueden ser falsos positivos o falsos negativos, aunque se han diagnosticado casos tan temprano como a las 14 semanas de gestación. Otras condiciones pueden simular una EPAR en el estudio ultrasonográfico, por lo que este estudio no es específico para el diagnóstico. A pesar de ello el ultrasonido prenatal puede aportar los datos siguientes: agrandamiento renal masivo, hiperecogenicidad del parénquima, no diferenciación córtico-medular, pequeños quistes menores de 2 cm de diámetro y aumento de la ecogenicidad hepática. El diagnóstico prenatal es posible utilizando análisis del ADN.21,27 En algunos casos el estudio en el período neonatal permite apreciar el gran tamaño de los riñones y los pequeños quistes (figura 1).
]]> Figura 1. Ultrasonido renal en un paciente de 26 días de nacido que muestra riñones muy aumentados de tamaño (9 cm el derecho y 8 cm el izquierdo) y pequeños quistes con diámetro entre 1,5 y 4,0 mm.
El estudio gammagráfico con ácido dimercaptosuccínico marcado con tecnecio 99 (Tc99m-DMSA) muestra captación en parches del radioisótopo, especialmente en los polos renales (figura 2).28
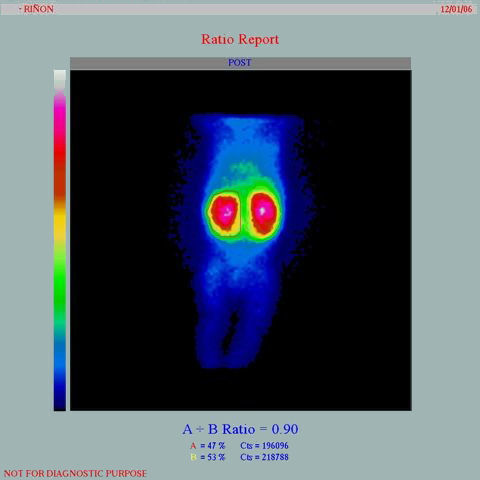
Figura 2. Estudio gammagráfico estático (DMSA), del mismo paciente de la figura 1, que muestra el gran tamaño de los riñones con hipocaptación bilateral de ambos polos, que se hace más evidente en el polo superior izquierdo en esta figura.
Todos los niños con EPAR tienen fibrosis hepática, por lo que la gammagrafía hepática con ácido metil-bromo-iminodiacético marcado con tecnecio 99 (Tc99m-HIDA) demostrará un lóbulo izquierdo agrandado con demora en el paso del contraste a través del hígado, en ocasiones con estancamiento y aumento prominente del sistema biliar. Este método no invasivo es de gran utilidad para el diagnóstico de EPAR.29
Aunque se ha señalado que la EPAR puede ser diferenciada de la enfermedad poliquística autosómica dominante (EPAD) sobre la base de sus características clínicas y radiográficas, puede existir un importante solapamiento tanto clínico como radiológico,30 que incluye la edad de comienzo de la sintomatología en ambos tipos, fibrosis hepática en la EPAD31 y la presencia de aneurismas cerebrales en la EPAR.32 Sin embargo, el diagnóstico correcto de EPAR debe descansar en la historia familiar con una forma de herencia recesiva y la biopsia hepática.33
El diagnóstico prenatal conlleva un pronóstico sombrío.23 Las muertes perinatales ocurren en los casos más graves y aunque la tasa de mortalidad perinatal no está bien establecida, un estudio entre 1974 y 1983 que incluye a 73 pacientes reportó 75 % de mortalidad en este período, 51 % en las primeras 24 horas, y en las necropsias se encontró hipoplasia pulmonar o neumotórax en 87 % de los casos.34
En un estudio de 91 niños diagnosticados a una media de edad de 2,3 meses y con masas renales palpables como manifestación más frecuente, en un período de observación de 4,1 años, de 39 pacientes con afectación renal funcional, 8 necesitaron tratamiento dialítico de mantenimiento y se trasplantaron 3. Este mismo estudio detectó una importante influencia del sexo y la evolución a largo plazo; la afectación de la función renal, el retardo del crecimiento y las infecciones urinarias fueron más frecuentes en el sexo femenino y los trastornos hepáticos predominaron en el varón.22 Se ha reportado una supervivencia de 86 % (29) y 87 % (7) al año de edad y 82 % a los 5 años.35
Más de la mitad de los pacientes con enfermedad renal poliquística autosómica recesiva/fibrosis hepática congénita (EPAR/FHC) requieren trasplante renal antes de alcanzar los 20 años de edad.7,8,34 Las principales causas de morbilidad en estos pacientes son: el inicio temprano de hipertensión arterial grave, insuficiencia renal, sangrado por várices esofágicas y colangitis recurrente,7,25,28,34 para lo cual sólo disponemos hasta el presente de tratamiento sintomático.26
]]> Resumiendo lo encontrado en un estudio reciente que reporta la evolución a largo plazo de 164 pacientes de 0 a 35 años, la tasa de supervivencia al año fue de 85 % y a los 10 años de 82 %, respectivamente; la insuficiencia renal se detectó por primera vez a los 4 años y la supervivencia renal fue de 86 % a los 5 años, 71 % a los 10 y 42 % a los 20. Aproximadamente 75 % de los pacientes desarrolló hipertensión arterial y 44 % presentó secuelas de hipertensión portal, y estas manifestaciones se correlacionaron con la edad.4La conducta médica ante la insuficiencia renal crónica de la EPAR es similar al manejo de la insuficiencia renal producida por otras causas. Sin embargo, la presencia de hipertensión y de defectos de concentración urinaria en la EPAR pueden requerir atención especial. La hipertensión en los pacientes con EPAR es tratada fundamentalmente con inhibidores de la enzima convertidora de angiotensina y bloqueadores de los receptores de angiotensina.26 También pueden utilizarse los bloqueadores de los canales del calcio, los beta-bloqueadores y juiciosamente los diuréticos de asa y las tiazidas.
No existe una guía estricta para la atención de los pacientes con síndrome de Caroli, con colangitis recurrente, sepsis y colelitiasis. Las derivaciones porto-sistémicas y el abordaje combinado de trasplante de hígado y riñón son muy complejos y deben individualizarse. Se reportan casos de trasplante hepático como opción terapéutica en que la función renal se ha mantenido estable a pesar de la utilización de ciclosporina A como terapéutica inmunosupresora.36 La disfunción esplénica que pueden presentar estos pacientes los expone al riesgo de infecciones por gérmenes encapsulados, por lo que los que presentan hipertensión portal pueden beneficiarse de la inmunización contra neumococos, meningococos y Haemophilus influenzae.26
Los niños afectados por las formas más graves de la enfermedad pueden sufrir dificultad respiratoria producida por las grandes masas renales que restringen la expansión del diafragma y la hipoplasia pulmonar asociada, pero si con las medidas de cuidados intensivos perinatales logran sobrevivir, pueden presentar dificultad en la alimentación enteral por la compresión digestiva. En estos casos se ha aplicado la nefrectomía paliativa unilateral o bilateral. Shukla y cols. reportan el caso de un lactante que sólo toleró la alimentación enteral después de una nefrectomía izquierda.23
Se están desarrollando protocolos de estudio en niños y adultos con diagnóstico de esta enfermedad hereditaria, que requieren la característica de la afectación renal y hepática típicas basada en la clínica y los estudios biópsicos con ultrasonido renal paterno normal e historia familiar compatible de herencia autosómica recesiva en busca de conocer mejor la enfermedad.26 Pero lo más necesario, pudiéramos decir imprescindible, en la EPAR es definir el defecto básico, incluyendo la función precisa de la proteína fibrocistina más completamente. Mientras no tengamos más elementos de las bases fisiopatológicas de la enfermedad, el tratamiento no dejará de ser sintomático.
Recessive autosomal and dominant autosomal polycystic kidney diseases are classically described as hereditary illnesses; they are also called polycystic disease of “child type” and of “adult type” respectively since both may be seen in any of these two life stages. The changing concepts of recessive autosomal disease, given the advances made in the treatment of newborns with this disease, and the location of the gen, the mutation of which causes it, encouraged us to make a brief literature review to help medical students and general practitioners to understand this disease.
Key words: recessive autosomal polycystic disease, congenital hepatic fibrosis, renal cysts.
1. Gagnadoux M-F, Broyer M. Polycystic kidney disease in children, En: Davison AM, Cameron JS, Grünfeld J-P, Keer DUS, Ritz S, Winearls CD, (Eds), The Oxford Textbook of Clinical Nephrology on CD-ROM. Chapter 15,2,1, Oxford University Press; 1997.
2. McDonald RA, Watkins SL, Avner ED. Polycystic kidney disease, En: Barratt TM, Avner ED, Harmon WE, (Eds). Pediatric Nephrology. 4th Ed., Baltimore: Lippincott William & Wilkins; 1998. Pp.459-464.
3. Álvarez V, Málaga S, Navarro M, Espinoza L, Hidalgo E, Badía J, et al. Analysis of chromosome 6p in Spanish families with recessive polycystic kidney disease. Pediatr Nephrol. 2000;14:205-207.
4. Bergmann C, Senderek J, Windelen E, Küpper F, Middeldorf I, Schneider F, et al. Clinical consequences of PKHD1mutations in 164 patients with autosomal recessive polycystic kidney disease (ARPKD). Kidney Int. 2005;57: 829-848.
5. Peters DJ, Losekoot M, de Die-Smulders CE, Stevens-Baldewijns M, Breuning MN. From gene to disease: PKHD1 and polycystic kidney disease, Ned Tijdschr Geneeskd. 2005;149: 463-466.
6. Harris PC, Rossetti S. Molecular genetics of autosomal recessive polycystic kidney disease. Mol Genet Metab. 2004;81:75-85.
7. Capisonda R, Phan V, Traubuci J, Daneman A, Balfe JW, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: Outcomes from a single-center experience. Pediatr Nephrol. 2003;18:119-126.
8. Adeva M, El-Youssef M, Rossetti S, Kamath PS, Kubly V, Consugar MB, et al. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine (Baltimore). 2006;85:1-21.
9. Gabow P, Grantham JJ. Polycystic kidney disease, En: Schrier RW, Gottschalk CW, (Eds). Diseases of the Kidney. 5th Ed, Volume I. Boston/Toronto/London: Little Brown & Company; 1992:535-556.
10. Ong AC, Wheatley DN. Polycystic kidney disease - The ciliary connection. Lancet. 2003;361:774-776.
11. Lambert P. Polycystic disease of the kidney. Arch Pathol. 1947;44:34-58.
12. Osathanond V, Potter E. Pathogenesis of polycystic kidneys: Type 1 due to hyperplasia of interstitial portions of collecting tubules. Arch Pathol. 1964;77.466-473.
13. Zerres K, Mucher G, Bachner L, Deschennes T, Eggermann T, Kääriäinen H, et al. Mapping of the gene for autosomal recessive polycystic kidney disease (ARPKD) to chromosome 6p21-cen. Nat Genet. 1994;7:429-432.
14. Blyth H, Ockenden BG. Polycystic disease of kidney and liver presenting in children. J Med Genet. 1971;8:257-284.
15. Sessa A, Righetti M, Battini G. Autosomal recessive and dominant polycystic kidney diseases. Minerva Urol Nefrol; 2004;56:329-338.
16. Consugar MB, Anderson SA, Rossetti S, Pankratz VS, Ward CJ, Torra R, et al. Haplotype analysis improves molecular diagnostics of autosomal recessive polycystic kidney disease. Am J Kidney Dis. 2005;45:77-87.
17. Reeders ST, Zhou J. The molecular genetics of inherited renal disease. En: Davison AM, Cameron JS, Grünfeld J-P, Keer DUS, Ritz S, Winearls CD, (Eds). The Oxford Textbook of Clinical Nephrology on CD-ROM. Chapter 16,1. Oxford University Press;1997.
18. Wilson PD. Polycystic kidney disease. N Engl J Med. 2004;350:151-164.
19. Torres VE, Harris PC. Mechanism of disease: Autosomal dominant and recessive polycystic kidney disease. Nat Clin Pract Nephrol. 2006;2:40-55.
20. Kogutt ME, Robichaux WH, Boineau FG, Drake GK, Simons CS. Asymetric renal size in autosomal recessive polycystic kidney disease. A unique presentation. AJR. 1993;160:835-836.
21. Bergmann C, Senderek J, Schneider F, Dornia C, Küpper F, Eggermann T, et al. PKHD1 mutations in families requesting prenatal diagnosis for autosomal recessivepolycystic kidney disease (ARPKD). Hum Mutat. 2004;23:487-495.
22. Zerres K, Deget F, Rudnik-Schoneborn S, Konrad M, and members of The Arbeitsgemeinschaft für Padiatrische Nephrologie. Bonn Heidelberg: Long-term Study of 91 children with autosomal recessive polycystic kidney disease (ARPKD): Clinical picture and influence of gender. Pediatr Nephrol. 1993;7:C18.F21 (Resumen).
23. Shukla AR, Kiddoo DA, Canning DA. Unilateral nephrectomy as palliative therapy in an infant with autosomal recessive polycystic disease. J Urol. 2004; 172: 2000-2001.
24. Barrera M, Quezada L. Riñón poliquístico infantil. Rev Chil Pediatr. 1986;57:35-38. (LILACS).
25. Summerfield JA, Nagafuchi Y, Sherlock S, Canafalch J, Schever PJ. Hepatobiliary fibropolycystic disease: A clinical and histological review of 51 Patients. J Hepatol. 1986;2:141-156.
26. Guynay-Aygun M, Avner ED, Bacallao RL, Choyke PL, Flynn JT, Germinio GC, et al. Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis: Summary statement of the First National Institute of Health/Office of Rare Disease Conference. J Pediatr. 2006;149.156-164.
27. Finer G, Birk O, Landan D. Genetic and phenotypic aspects of autosomal recessive polycystic kidney disease in Southern Israel. Harefuah. 2004; 143:466-470. (MEDLINE).
28. Roy S, Dillon MJ, Trompeter RS, Barratt TM. Autosomal recessive polycystic kidney disease: Long-term outcome of neonatal survivors. Pediatr Nephrol. 1997;11:301-306.
29. Gordon I, de Bruyn A. Diagnostic imaging. En: Barratt TM, Avner ED, Harmon WE, (Rds). Pediatric Nephrology. 4th Ed, Chapter 22. Baltimore: Lippincott William & Wilkins; 1998. Pp.377-390.
30. McDonald RA, Avner ED. Inherited polycystic kidney disease in children. Semin Nephrol. 1991;11:632-642.
31. Cobben JM, Breuning MH, Schoots G. Congenital hepatic fibrosis in autosomal dominant polycystic kidney disease. Kidney Int. 1990;38:880-885.
32. Neumann K, Krumme B, van Velthoven V. Multiple intracranial aneurysms in a patient with autosomal recessive polycystic kidney disease. Nephrol Dial Transplant. 1999;14:936-939.
33. Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. Renal cystic diseases: A review. Adv Anat Pathol. 2006;13:26-56.
34. Zerres K, Rudnik-Schoneborn S, Deget F, Holckamp U, Brodehl J, Geisert J, et al. Clinical course of 115 children with autosomal recessive polycystickidney disease. Acta Pediatr. 1996;85:437-445.
35. Kaplan BS, Fay J, Dillon M, Barratt TM. Autosomal recessive polycystic kidney disease. Pediatr Nephrol. 1989;3:43-49.
37. Arikan C, Ozgenc F, Akman SA, Kilic M, Tokat Y, Yagci RV, et al. Impact of liver transplantation on renal f unction of patients with congenital hepatic fibrosis associated with autosomic recessive polycystic kidney disease. Pediatr Transplant. 2004;8:558-560.
]]>
1 Especialista de II Grado en Pediatría. Profesor Consultante de Pediatría.