Dr. Iván Hernández García,1 Dra. Mirely Fernández Martín,2 Dra. Silvia León Pérez,3 Dra. Alina García García4 y Jorge Riaño Echenique5
Resumen
La osteogénesis imperfecta clasifica entre las displasias óseas por alteraciones en la densidad y los defectos del modelaje óseo. El tipo I es la forma más frecuente de la enfermedad y se caracteriza por un patrón de herencia autosómico dominante. No es infrecuente que la enfermedad aparezca producto de una nueva mutación. También se ha demostrado que puede ser producida por mosaicismos germinales. Este trabajo documenta, por primera vez en Cuba, el caso de una familia con 3 individuos de diferente sexo afectados por osteogénesis imperfecta de tipo I mientras ninguno de los progenitores lo está. Se discute la posibilidad etiológica de un mosaicismo germinal y se valora asimismo la posibilidad de un patrón de herencia distinto del dominante, lo cual aportaría nueva evidencia de heterogeneidad genética.
Palabras clave: Osteogénesis imperfecta, mosaicismo germinal, heterogeneidad genética.
El término osteogénesis imperfecta (OI) designa una enfermedad que tiene entre sus características más sobresalientes la susceptibilidad a las fracturas y las deformaciones óseas. La clasificación más comúnmente utilizada divide a los pacientes en cuatro tipos (I-IV).1 El mejor conocimiento de las bases moleculares de la enfermedad y el uso de novedosos enfoques diagnósticos, entre los que se encuentra la histomorfometría ósea, ha permitido refinar la clasificación añadiendo nuevos grupos. Los tipos V, VI, y VII fueron identificados entre aquellos que presentaban el tipo IV, basado en las anomalías óseas y la ausencia de mutaciones en los genes COL1A1 y COL1A2 y no está claro si deban agruparse junto a la OI en el futuro.2-4
La OI de tipo I es la forma más frecuente de la enfermedad (1:30 000). La mutación más comúnmente encontrada se produce en el gen COL1A2 que codifica para la cadena alfa2 del procolágeno1 y produce disminución del colágeno de tipo 1, que es estructuralmente normal. Se hereda de forma autosómica dominante. No es infrecuente, sin embargo, que la enfermedad aparezca producto de una mutación de novo (60 % para el tipo I).5 Se ha demostrado también que puede aparecer como resultado de mosaicismos germinales (MG) y somáticos.6,7
En este trabajo se describe a una familia donde se encuentran 3 individuos de diferente sexo afectados por OI de tipo I, mientras ninguno de los progenitores la padecen. Por la importancia que tiene el conocimiento del patrón de transmisión de la enfermedad, que se ha demostrado heterogéneo, y de los riesgos de recurrencia, y considerando que no tenemos conocimiento de otro reporte similar en nuestro país, hemos decidido presentar esta familia afectada muy probablemente por mosaicismo germinal y hacer una breve revisión sobre el tema.
]]>Se incluyen en este trabajo 3 individuos con características clínicas compatibles con el diagnóstico de OI de tipo I. Dos sujetos del sexo femenino, de 17 y 22 años respectivamente, y un varón de 20 años de edad. Ninguno había sido valorado antes por especialistas en Genética Clínica. Se les confeccionó un detallado árbol genealógico y se hizo un exhaustivo examen físico y un interrogatorio minucioso para evidenciar antecedentes familiares de la enfermedad.
La familia procede del municipio Rodas, en la provincia de Cienfuegos. El padre tiene 47 años y la madre 43; ambos son de la raza negra. Fueron entrevistados en el hogar en relación con antecedentes patológicos personales o familiares de la enfermedad, los que fueron negativos. No encontramos defectos físicos que indicaran secuelas de fracturas ni alteraciones en la dentina y la esclerótica que sugirieran expresión mínima de la enfermedad o mosaicismo somático. No son consanguíneos.
Caso 1. Paciente del sexo femenino, de 22 años de edad. Nacida de parto eutócico luego de un embarazo a término, con peso adecuado y desarrollo psicomotor normal. Refiere antecedentes de salud normal hasta 2 y medio años, fecha en que comienza a presentar múltiples fracturas en clavículas y huesos de la mano. Durante toda la infancia se suman fracturas de muñeca, antebrazo, fémur, tibia y peroné, y huesos de la pelvis.
Al examen físico constatamos baja talla (140 cm). Se observan cicatrices amplias en los miembros inferiores, secuelas de múltiples fracturas. Presenta acortamiento del miembro inferior derecho, pie plano y metatarso varo. En radiografías (RX) previas se constatan fracturas y deformidades, así como disminución de la densidad ósea. Las escleras y los dientes eran normales al momento de la inspección. No se encontraron trastornos auditivos. Desde hace varios años se encuentra libre de fracturas. Tiene una niña de 17 meses de edad, que ha sufrido recientemente fractura de cubito y radio derechos.
Caso 2. Paciente del sexo masculino, de 20 años de edad. Nacido producto de embarazo a término y parto normal. Comenzó a presentar fracturas de miembros superiores e inferiores y clavícula desde los 13 meses de edad. Al examen físico se constata ligera incurvación del miembro inferior derecho. Presenta estatura normal (165 cm), pie plano y metatarso varo. Las escleras y los dientes no presentan anomalías al examen visual. Se constatan fracturas antiguas en los RX. Ha presentado una prolongada remisión de la enfermedad desde los 4 años de edad.
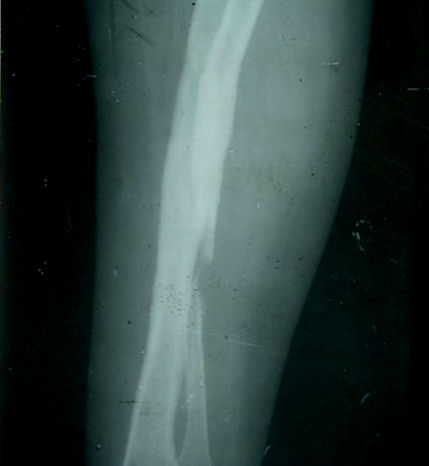
Caso 3. Paciente del sexo femenino, de 17 años de edad, nacida de parto eutócico con peso y desarrollo psicomotor normal. Presentó fractura de ambas clavículas al momento del parto. A partir de los 4 meses de edad continuó presentando fracturas recurrentes ante pequeños traumatismos. Se constata baja talla (143 cm). Se observan grandes deformidades con cicatrices amplias producto de operaciones correctivas, sobre todo en el miembro superior derecho. Se constató genu valgo, pie plano y metatarso varo bilateral e hiperlaxitud articular de la muñeca y las rodillas (genu recurvatum). Se confirman en los RX fracturas de cúbito y radio, en el antebrazo derecho ausencia de un segmento del hueso debido a secuestros óseos y deformación concomitante, que ha requerido varias intervenciones quirúrgicas (figura). Las escleras y los dientes son normales. No hallamos trastornos auditivos. Hace un año que no presenta fracturas.
]]>Figura. Radiografía correspondiente al paciente del caso 3.
Osteogénesis imperfecta de tipo I no es sinónimo de OI ligera. Los pacientes frecuentemente tienen estatura normal. Pueden tener pocas o ninguna fractura, la mayor parte durante de los primeros años de vida o incluso al nacimiento o numerosas fracturas a lo largo de sus vidas.6,7 Por lo general pueden valerse por sí mismos completamente. Las escleras son azules, aunque en ocasiones va disminuyendo el tono azuloso y en la pubertad o adultez temprana pueden verse completamente blancas.
No existe consenso acerca de las características básicas de las diferentes formas clínicas. Por ejemplo, no está claro si este tipo incluye individuos con baja estatura o si la deformidad descarta el diagnóstico de OI de tipo I. Hemos descartado en el interrogatorio y examen físico una forma benigna en ambos progenitores. En el caso del padre, es muy improbable ya que siempre ha trabajado en la construcción y por tanto está expuesto a numerosos traumatismos que, sin embargo, jamás le han provocado fractura.
La presencia de varios tipos de OI y de diferentes patrones de herencia, incluidos los no clásicos, pone de manifiesto la gran heterogeneidad genética de la enfermedad. Aunque para el tipo I no se tiene reporte de un patrón de herencia diferente del dominante, no hay evidencia alguna de consanguinidad entre los padres y según el modelo bayesiano la probabilidad de que, siendo ambos padres portadores de una mutación recesiva nazcan 3 hijos afectados, es de 1 en 15 625 o de 0,000064 %, la que resulta extremadamente baja. Sin embargo la prueba más importante de que en este caso no se debe a una mutación de tipo recesiva la aporta el inicio reciente de fracturas que ha tenido la hija de uno de los afectados. .
La osteogénesis imperfecta de tipo II tiene una incidencia de 1-2 por 100 000 habitantes. En esta forma la mayoría de los recién nacidos no sobrevive al período perinatal. Las causas de muerte son fundamentalmente las malformaciones y hemorragias del sistema nervioso central, la fragilidad de las costillas y la hipoplasia pulmonar. Los infantes presentan múltiples fracturas intrauterinas, incluyendo el cráneo, huesos largos y vértebras, y graves deformidades de los huesos largos.8 La mayoría de los casos se debe a nuevas mutaciones autosómicas dominantes.9,10
Se ha descrito el caso de OI letal de un paciente cuyo padre era portador de un mosaicismo germinal y somático con muy poca repercusión clínica. El estudio molecular arrojó idéntica proporción del gen COL1A2, normal y con mutación.6 Hay autores que ponen en duda la existencia del tipo de herencia autosómico recesivo en esta forma clínica.
La osteogénesis imperfecta de tipo III (OI grave) registra uma incidencia de 1-2 por 100 000. Estos pacientes tienen una facies descrita como triangular producto del sobrecrecimiento de la cabeza y del poco desarrollo de los huesos de la cara.2,3 Existen graves deformidades torácicas y de huesos largos, baja talla marcada, además de fracturas vertebrales y escoliosis. Generalmente los pacientes se encuentran confinados a una silla de ruedas. Las escleras son azules en la infancia y la sordera comienza en la juventud. El diagnóstico prenatal es difícil pero posible por medio de ultrasonografía.11 Muchos casos pueden tener complicaciones vertebrales que comprometen la supervivencia. La pérdida auditiva es frecuente. Se describe herencia dominante y patrones recesivos muy raramente. Por las características clínicas de nuestros pacientes podemos descartar esta forma.
La osteogénesis imperfecta de tipo IV es probablemente tan común como la OI de tipo I. Son individuos con OI moderada y baja estatura, pero tienen además angulación de los huesos largos y fracturas vertebrales. Pueden estar presentes la escoliosis y la laxitud articular. Los pacientes con este tipo de OI generalmente son capaces de deambular, pero requieren ayuda para caminar. Basado en la presencia o no de dentinogénesis imperfecta (DI) se ha subdividido este tipo en las formas ‘a’ y ‘b’.2,12 Ambas formas tienen escleras normales. Es la forma más variable de OI.
La DI es común pero ligera. Su diagnóstico preciso es frecuentemente difícil debido a que las características clínicas no están bien delineadas en la literatura especializada. Aunque dos de nuestros pacientes presentan ligera angulación de sus huesos largos, el cuadro clínico y el hecho de que podían valerse por sí mismos nos hacen descartar este tipo de OI.
]]> La osteogénesis imperfecta de tipo V se confundió en un inicio con las formas III y IV debido a la baja talla y la naturaleza de las fracturas. Las escleras son generalmente normales. Dos hallazgos lo distinguen especialmente: la formación de un callo hipertrófico en el sitio de las fracturas y la calcificación de la membrana interósea entre el cúbito y el radio que lleva a la incapacidad de supinar y pronar completamente el antebrazo.5 No se detectan anomalías del colágeno en las biopsias. Se hereda con un carácter autonómico dominante.Los hallazgos clínicos en la osteogénesis imperfecta de tipo VI son similares a los de la OI de tipo IV, como la baja talla y la angulación de los huesos largos. La biopsia del hueso es necesaria en el diagnóstico de los tipos V y VI, y en este último muestra un defecto grave de la mineralización, que lo distingue del resto de las formas clínicas. En este grupo tampoco se detectan anomalías del colágeno. Se desconoce su patrón de herencia. Los tipos V y VI se estima constituyen menos del 5 % de los casos de OI.4,5
La osteogénesis imperfecta de tipo VII se ha encontrado hasta la fecha solo en poblaciones nativas de Canadá, específicamente en la provincia de Québec. Se distingue por el acortamiento rizomélico de todos los miembros y es heredada de forma autosómica recesiva.2,5
Ninguno de los tipos V, VI o VII tiene una mutación identificable en los genes COL1A1 o COL1A2.
El mosaicismo germinal (MG) ha sido reconocido recientemente entre las causas frecuentes de enfermedad genética.13 Hay dos posibilidades para la existencia de los mosaicismos. Una, en la cual la mutación ocurre en una célula germinal que está en división. La otra, cuando la mutación ocurre muy temprano en una célula somática, antes de la separación de ésta en célula germinal y por ende presente en ambos tipos celulares. En dependencia del gen involucrado y el grado de mosaicismo, el individuo portador de una mutación somática y germinal podría ser asintomático o presentar signos y síntomas de la enfermedad. Todavía se encuentran pocos reportes en la literatura donde se analicen los MG.
Entre las enfermedades en las que se ha reconocido se encuentra la neurofibromatosis de tipo I y la OI.6,14 Debe sospecharse MG en las familias con enfermedades genéticas de herencia autosómica dominante y si existe más de un hijo afectado de padres sanos. En la literatura especializada se recoge un caso de OI autosómica dominante mortal, en el cual ambos padres son mosaicos para la línea germinal y somática de una mutación en el gen COL1A2.6
En los casos de MG, el número de sujetos con OI de un mismo árbol genealógico es extremadamente variable, ya que estará en relación con la proporción de células afectadas y esto depende del momento de la división de las células sexuales en que ocurre la mutación. Los individuos en los cuales la mutación ha ocurrido en la primera generación de las células primordiales –que dan origen a las gónadas–, el 100 % de los gametos estarán afectados; la mitad de estos si la mutación ocurre en la primera división de estas células y, en general, 1/2 n si la mutación ocurre en cualquiera de las n divisiones. El mosaicismo para mutaciones dominantes ha sido reconocido en la OI de tipo II, III y V.5,7
El fenotipo de los individuos mosaicos puede variar desde características no identificables hasta formas ligeras de la enfermedad. Los individuos mosaicos para mutaciones que resultan en formas no letales de OI generalmente no tienen hallazgos fenotípicos de OI incluso cuando la mutación esté presente en la mayoría de las células del organismo. El mosaicismo de mutaciones que resultan en OI letal puede producir una OI ligera si la mutación está presente en la mayoría de las células somáticas.
Diagnóstico prenatal
El diagnóstico prenatal (DPN) de embarazos en riesgo de OI debe ser llevado a cabo analizando el colágeno sintetizado en las células fetales obtenidas por biopsia de vellosidades coriales, entre las semanas 10 a 12 de la gestación, si se ha demostrado una anomalía en el colágeno de células cultivadas del probando. Debido a que los amniocitos no producen colágeno de tipo I, no es posible hacer el DPN utilizando esta estirpe celular.5
]]> El DPN también puede ser realizado por el estudio molecular de los genes COL1A1 y COL1A2, si la mutación ha sido identificada en uno de los familiares afectados. Ninguno de estos dos estudios mencionados se lleva a cabo aún en nuestro país. Un examen ultrasonográfico realizado en un centro de experiencia puede ser de utilidad en el diagnóstico de las formas más graves, antes de las 20 semanas de gestación. Los fetos afectos por formas ligeras pueden ser detectados tardíamente en el embarazo cuando ocurren las fracturas y deformidades.
Osteogenesis imperfecta is one of the bone dysplasias caused by altered density and bone model defects. Type I is the most common form of disease and is characterized by an autosomal dominant inheritance pattern. Sometimes, this disease occurs as a result of a new mutation. It has been also demonstrated that it can be caused by germ mosaicisms. This paper documented for the first time in Cuba the case of a family with three (3) individuals of both sexes affected by type-1 osteogenesis imperfecta but their parents were not. The etiological possibilities of germ mosaicism and the possibilities of an inheritance pattern different from the dominant one were discussed, which would give new genetic heterogeneity evidence.
Key words: osteogenesis imperfecta, germ mosaicism, genetic heterogeneity.
1. Sillence DO, Senn A, Danks DM. Genetic heterogeneity in Osteogenesis Imperfecta. J Med Genet. 1979;16:101–16.
2. Plotkin H, Primorac D, Rowe, D. Osteogenesis Imperfecta. In: Glorieux F, Pettifor J, Juppner J. Pediatric Bone – Biology and Disease. San Diego, CA: Elsevier Science; 2003. Pp. 443–471.
3. Glorieux FH, Wart L, Rauch F, Lalic L, Roughley P, Travers R. Osteogenesis Imperfecta Type VI: a form of brittle bone disease with a mineralization defect. Journal of Bone and Mineral Research. 2002;17:30–38.
4. Glorieux FH, Rauch F, Plotkin H, Wart L, Travers R, Roughley P, et al. Type V Osteogenesis Imperfecta: a new form of brittle bone disease. J Bone Miner Res. 2000;15:1650–1658.
5. Gene reviews database. [en línea] Consultado el 25 de diciembre de 2006. Disponible en: http://www.geneclinics.org/profiles/all.html
6. Edwards M, Wenstrup R, Byers P, Cohn D. Recurrence of lethal Osteogenesis Imperfecta due to parental mosaicism for a mutation in the COL1A2 gene of type I collagen. The mosaic parent exhibits phenotypic features of a mild form of the disease. Hum Mutat. 1992;1(1):47-54.
7. Cabral Wayne A, Marini Joan C. High proportion of mutant osteoblasts is compatible with normal skeletal function in mosaic carriers of Osteogenesis Imperfecta. Am J Hum Genet. 2004;74(4):752-60.
8. Plotkin H, Lutz R. Osteoporosis in pediatrics. In: Deng HW, Liu Y. Osteoporosis: Basic and Clinical Aspects. Omaha: World Scientific Publishing; 2004.
9. Shapiro JR, Burn VE, Chipman SD, Jacobs JB, Schloo B, Reid L, et al. Pulmonary hypoplasia and Osteogenesis Imperfecta type II with defective synthesis of alpha I(1) procollagen. Bone. 1989;10:165–171.
10. Cole WG, Dalgleish R. Perinatal lethal Osteogenesis Imperfecta. J Med Genet. 1995;32:284–289.
11. Robinson LP, Worthen NJ, Lachman RS, Adomian GE, Rimoin DL. Prenatal diagnosis of Osteogenesis Imperfecta type III. Prenatal Diagnosis. 1987;7:7–15.
12. Levin LS, Salinas CF, Jorgenson RJ. Classification of Osteogenesis Imperfecta by dental characteristics [letter]. Lancet. 1978;1:332–3.
13. Zlotogora. D. Germ line mosaicism. Hum Genet. 1998 Apr;102(4):381-6.
14. Lázaro C, Gaona A, Lynch. M, Kruyer H, Ravella A, Estivill X. Molecular characterization of the breakpoints of a 12-kb deletion in the NF1 gene in a family showing germ-line mosaicism. Am J Hum Genet. 1995;57(5):1044-9.
]]> Recibido: 2 de febrero de 2007. Aprobado: 15 de mayo de 2007.
1 Especialista de I Grado en Genética Clínica. Instructor del Hospital Pediátrico Docente «William Soler».
2 Especialista en Medicina General Integral. Máster en Asesoramiento Genético. Policlínico Docente «Felipe Poey Aloy», Nueva Paz.
3 Especialista de I Grado en Pediatría. Instructora del Policlínico Docente «Felipe Poey Aloy», Nueva Paz.
4 Especialista de I Grado en MGI. Especialista de I Grado en Genética Clínica. Instructora del Hospital Pediátrico Docente «William Soler».
5 Especialista de II Grado en Ortopedia. Profesor Auxiliar del Hospital Pediátrico Docente «William Soler». ]]>