Dra. Yoerquis Mejías Sánchez,1 Dr. Orgel José Duany Machado2 y Dr. Noel Taboada Lugo3
Resumen
Los trastornos de la diferenciación sexual constituyen un grupo complejo de entidades y síndromes. El término estados intersexuales hace referencia a aquellos recién nacidos que presentan genitales ambiguos, esto es, sin evidencia clara sobre sexo asignable. Su frecuencia en nuestro medio es relativamente escasa. Se presenta el caso de un recién nacido en el que, en el examen físico, presenta micropenisomía y escroto bífido. La fusión labioescrotal está alterada con rodetes separados que semejan labios mayores y una estructura que remeda un introito vaginal. En el estudio ultrasonográfico se descartó la presencia de útero y ovarios y se constató la presencia de testículos en canal inguinal. La cromatina sexual de células en interfase de la mucosa oral reveló la presencia de 0 % de cuerpo Barr y el estudio cromosómico mostró un cariotipo masculino normal (46, XY), por lo que se interpreta el caso como un pseudohermafroditismo masculino. Se realiza una revisión de los trastornos de la diferenciación sexual.
Palabras clave: Diferenciación sexual, trastornos, genitales ambiguos, hermafroditismo, pseudohermafroditismo, disgenesia gonadal mixta.
Cuando nace un niño con sexo incierto; calamidad y aflicción
se apoderan del lugar, y el señor de la casa nunca será feliz.
Proverbio babilónico (1700 a.n.e)
En nuestros días este antiguo proverbio mantiene su vigencia, pues la intersexualidad plantea un delicado problema que es necesario manejar con mucho cuidado para evitar desagradables consecuencias. La intersexualidad queda definida en sentido amplio por la existencia de cualquier discordancia entre los criterios de definición: sexo cromosómico, gonadal, genital, fenotípico o morfológico y psicosocial.1
El sexo va a ser definido, primeramente a nivel genético, por el establecimiento de los cromosomas sexuales que se encuentren en el cigoto. Esta expresión genética lleva a que las estructuras no diferenciadas se desarrollen en el tipo de gónadas que corresponde según el estímulo que reciban, para alcanzar finalmente el sexo fenotípico. Entre las cuatro y seis semanas de desarrollo embrionario en ambos sexos, las células germinales primordiales migran desde su localización extraembrionaria primitiva en el mesodermo hasta la protuberancia gonadal, donde son rodeadas por los cordones sexuales para formar un par de gónadas primitivas. Hasta este momento las gónadas en desarrollo, con independencia de la constitución cromosómica femenina o masculina, es bipotencial y con frecuencia se le refiere como «indiferente».2
]]> El desarrollo hacia ovarios o testículos está determinado por la acción coordinada de una secuencia de genes que conllevan a la diferenciación gonadal femenina cuando no existe el cromosoma Y, o el desarrollo testicular cuando este cromosoma está presente. En 1987 Affara y cols. prueban con sondas complementarias a la región del brazo corto de este cromosoma y el ADN de individuos, que eran fenotípicamente varones pero que a nivel citogenético presentaban cromosomas sexuales XX, y descubren que dichas sondas hibridizan, es decir que eran complementarias al ADN de estos individuos. Este resultado los llevó a deducir que existía material genético del brazo corto del cromosoma Y en estos individuos, y se logró establecer una región responsable del desarrollo testicular en el brazo corto del cromosoma Y (Yp). Más tarde, Page determina que en esta región se ubica un gen al que se llamó SRY (del inglés ‘sex-determining region on the chromosome Y’), que codifica para un factor de transcripción que actúa a modo de interruptor e inicia la cascada de desarrollo gonadal masculino.En los estadios embrionarios iniciales los genitales externos consisten en un tubérculo genital, un par de pliegues uretrales y otros labioescrotales también pareados. En el sexo masculino (cariotipo 46, XY) las células de Leydig de los testículos fetales producen andrógenos, que estimulan a los conductos mesonéfricos (antes llamados Wolffianos) a formar pene, bolsas escrotales, cordón y vesículas seminales, y las células de Sertoli producen una hormona (sustancia inhibitoria mülleriana) que suprime la formación de los conductos paramesonéfricos (antes conocidos como müllerianos). Así, a partir de ese estado indiferenciado, los genitales externos masculinos se desarrollan. Cuando el gen SRY no está presente, la gónada se convierte en un ovario (lo que indica sexo femenino; cariotipo 46, XX) con involución de los conductos mesonéfricos y los paramesonéfricos que forman útero y trompas de Falopio.1,2
Además del mencionado SRY, otros genes en el cromosoma X y en algunos autosomas están implicados en la conversión de esa gónada indiferenciada en testículos u ovarios. El gen DAX1 ubicado en el brazo corto del cromosoma X (Xp21) está involucrado en una interacción de regulación con el SRY en un período crítico del desarrollo, que conlleva a la formación de testículos. Así, una duplicación de este gen resulta en un exceso del factor de transcripción codificado, lo que puede suprimir la función determinante masculina normal del gen SRY, y resultar en un desarrollo ovárico.
El gen SOX9, ubicado en el brazo largo del cromosoma 17, se expresa en períodos iniciales del desarrollo y es requerido para la formación normal de los testículos. También los genes WT1 y DMRT1 con loci en los brazos cortos de los cromosomas 11 y 9 respectivamente, codifican para factores de transcripción relacionados con el desarrollo gonadal. Mutaciones dominantes en estos genes alteran el desarrollo testicular normal.3
La determinación del sexo y asignación de un género es muy difícil en algunos recién nacidos porque los genitales son ambiguos. Cuando existe una ambigüedad genital se emplea el término de estado intersexual o intersexos. En algunos pacientes están presentes tanto tejido ovárico como testicular, condición conocida como «hermafroditismo». El término «pseudohermafroditismo», por su parte, se reserva a individuos con tejido gonadal de un solo sexo.1,3 El origen mitológico del nombre proviene de la diosa Afrodita: el hijo de Hermes y Afrodita se bañaba en una fuente de Halicarnaso cuando la ninfa protectora de ésta, se le abrazó enamorada y suplicó a los dioses que la fundieran con él. Su plegaria fue escuchada y quedaron unidos en un solo cuerpo.4
La importancia de los estados intersexuales es evidente por la necesidad de realizar el diagnóstico tempranamente para su tratamiento precoz y por la trascendencia que la definición del sexo tiene para el futuro del individuo.
Paciente nacido a las 41,3 semanas de gestación producto de un parto eutócico, líquido claro, sin sufrimiento fetal agudo (Apgar 9/9). Las mensuraciones estuvieron dentro de parámetros normales: peso 3750 g; talla 52 cm; perímetro cefálico 37 cm y perímetro torácico de 36 cm. Edad materna al embarazo: 26 años.
Historia obstétrica: 5 gestaciones, 2 partos anteriores (una hembra y un varón fenotípicamente sanos), 2 abortos (interrupciones provocadas del embarazo por decisión de la pareja). No se recogen antecedentes de trastornos genéticos ni de otra índole, tanto por vía materna como paterna. Al revisar el carné obstétrico se constata que se trató de un embarazo normal, clasificado como de bajo riesgo obstétrico, con un adecuado seguimiento médico. Todos los exámenes de laboratorio realizados en los tres trimestres mostraron resultados compatibles con la normalidad.
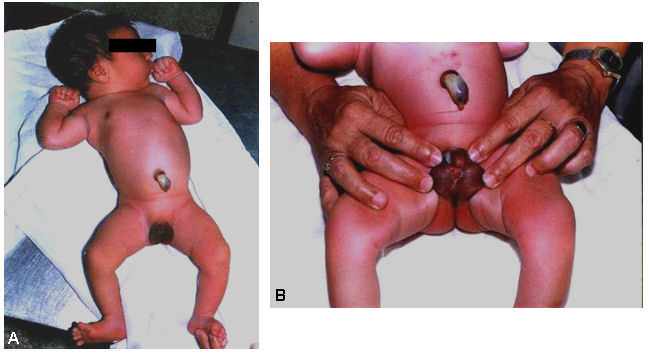
En el examen dismorfológico realizado al nacimiento se encontraron hallazgos positivos solamente a nivel de los genitales externos: micropenisomía y engrosamiento a lo largo de la línea media en su cara ventral, dado por una hiperplasia del rafe desde la base del pene hasta el prepucio, con un orificio uretral anatómicamente bien situado en el extremo del glande. Por debajo del micropene se encuentran dos rodetes cutáneos que remedan labios mayores y que al separarlos hacen evidente una estructura que simula un introito vaginal, o bien unos escrotos bífidos e hipoplásicos (figura).
]]>
Figura. A) Aspecto general del recién nacido. B) Aspecto morfológico de los genitales externos.
Exámenes complementarios:
Dada la utilidad de conocer los niveles plasmáticos de testosterona y cetoesteroides en este caso, se remitió al paciente al servicio de endocrinología para cuantificación y posterior seguimiento. La impresión diagnóstica es de pseudohermafroditismo masculino.
]]>A pesar de que la mayoría de los niños nacen normales, variadas situaciones genéticas pueden conducir, en algunos casos, a que presenten genitales ambiguos ante los cuales ni doctores ni familia están seguros del sexo del recién nacido. Este tipo de trastorno puede representar una experiencia traumática para los padres. Los bebés intersexuales se presentan aproximadamente en uno de cada 1000 nacimientos.5
Todo paciente con una anomalía de la diferenciación sexual debe ser tratado por un equipo especializado, que incluye el tratamiento psicológico adecuado a los padres y familiares. Debe ser valorado por ginecoobstetras, urólogos, pediatras, endocrinólogos, genetistas y psicólogos.
Al médico que examine al niño por primera vez corresponde una orientación correcta que debe quedar limitada a hablar con los padres del paciente refiriéndoles que su bebé presenta un defecto congénito, es decir un defecto desde el nacimiento, que afecta los genitales externos como en otros afecta al corazón. Este defecto que provoca que no haya completado su desarrollo genital y necesita ser visto por otros especialistas. Se les debe explicar a los padres que el bebé debe tener un desarrollo psicosexual normal, para lo cual debe ser estudiado por varios especialistas para la posterior asignación de un género adecuado, y de ser necesario, para instaurar tratamiento desde la más temprana edad.1
Los trastornos de la diferenciación sexual constituyen un grupo complejo de entidades y síndromes. Los estados intersexuales pueden ser incluidos dentro de los siguientes grupos:
La correcta diferenciación entre hermafroditismo verdadero y DGM resulta de gran importancia, porque en aquellos individuos con este último trastorno se recomienda una gonadectomía bilateral tan pronto como sea posible, ya que la gónada disgenésica tiene un alto potencial de malignización (aproximadamente un tercio de los pacientes desarrolla un gonadoblastoma durante la primera a cuarta década de la vida) y además a causa de que el 30 % de los gonadoblastomas se superponen con tumor de células germinales malignas como germinoma, tumor del seno endodérmico (tumor del saco vitelino), teratoma inmaduro, carcinoma embrionario o coriocarcinoma.
]]> En el pseudohermafroditismo masculino el cariotipo es 46, XY pero los genitales externos están incompletamente virilizados, son ambiguos o completamente femeninos. Cuando es posible encontrar las gónadas, éstas son testículos cuyo desarrollo morfológico puede oscilar desde rudimentario hasta normal. Existen dos causas principales para el pseudohermafroditismo masculino:La enzima 5-alfa reductasa convierte la testosterona en dihidrotestosterona (DHT), necesaria para la masculinización final del feto varón. Esta deficiencia se hereda por medio de un gen autosómico recesivo, lo que significa que cada padre es portador de una copia del gen mutado y a su vez lo transmite a su hijo. Los padres portadores tienen, en cada embarazo, un 12,5 % de posibilidades (una posibilidad en ocho) de tener un niño afectado, ya que esta deficiencia sólo afecta a los varones. Aunque el desarrollo testicular es normal, el pene es muy pequeño, y puede haber un saco vaginal ciego. Casi siempre los pacientes son criados como niñas, pero en la pubertad, la producción de testosterona por los testículos permite el desarrollo masculino, que se traduce en engrosamiento de la voz y aumento del tamaño del pene y de la masa muscular. No obstante, los órganos que responden normalmente a la dihidrotestosterona (escroto, testículos, próstata) continúan siendo prepuberales debido a la deficiencia de este esteroide.3,5-7 La mayor incidencia de estos casos a nivel mundial se reporta en la comunidad Las Salinas», al sur de República Dominicana. Únicamente Nueva Guinea tiene tantos ejemplos de esta alteración genética.8
Se denomina «síndrome de insensibilidad andrógena» o síndrome de Morris (antes llamado «feminización testicular») al trastorno genético que provoca que los bebés varones no respondan a los andrógenos (testosterona). Éste es un síndrome que se produce por una mutación en el gen receptor de andrógeno en el cromosoma X, por lo cual su herencia se describe como «recesiva ligada al X». Las madres portadoras del gen tienen un 50 % de posibilidades de tener hijos con síndrome de insensibilidad andrógena o hijas con un 50 % de posibilidades de ser portadoras de la mutación. Las personas afectadas son cromosómicamente masculinas (46, XY) con genitales externos aparentemente femeninos, con vagina ciega, sin útero ni ovarios. Los testículos están presentes en el abdomen o en el canal inguinal, y muchas veces son confundidos con hernias en los niños con apariencia fenotípica femenina normal.
El pseudohermafroditismo femenino, a su vez, puede ser causado por:
Existe otro tipo de HAC, denominada «perdedora de sal» que puede ser muy grave, incluso mortal, ya que se puede producir un colapso electrolítico en el recién nacido. Existen tratamientos disponibles si se diagnostica a tiempo. Los hombres y las mujeres se ven afectados por igual. Existen otros problemas de enzimas menos comunes, que pueden derivar en HAC, tanto en hombres como en mujeres.
El tamaño normal del pene del recién nacido es de 3,5 ± 0,7 cms. La micropenisomía puede resultar de un fallo testicular primario o secundario durante la vida fetal, después de que la morfogénesis es completa5 y puede ocurrir en niños con algunos síndromes genéticos como el de Ulrich-Noonan, Robinow, Carpenter, Cornelia de lange, Down, anemia de Fanconi, Hallerman-Streiff, y deleción del brazo largo del cromosoma 18 (18q-). También puede verse asociado en niños con hipoglicemia debido a hipopituitarismo.3,9
Para un adecuado enfoque del hipogonadismo es útil diferenciar entre la falla testicular (hipogonadismo primario o hipergonadotrópico) y los trastornos del eje hipotálamo-hipófisis-testículo (EHHT), que corresponden al hipogonadismo secundario, también denominado hipogonadotrópico. La deficiencia primaria de la gónada masculina puede presentarse como parte integral de varios síndromes, tales como el síndrome de Klinefelter, el síndrome de Reifenstein y el síndrome de Ulrich-Noonan, o aparecer a consecuencia de destrucción del testículo (por trauma, compromiso vascular o tuberculosis), enfermedades sistémicas o autoinmunitarias y anorquia. Por su parte, el rasgo distintivo del hipogonadismo secundario es la deficiencia de gonadotropinas y es el resultado de daño de la hipófisis o del área tuberal del hipotálamo, donde están localizadas las células productoras de hormona liberadora de gonadotropinas (HLGn), como se ve en el síndrome de Kallman, Prader Willi, y el de Rud, entre otros.10,11
El tratamiento para los trastornos de la diferenciación sexual dependerá de su tipo específico, pero se suele realizar una cirugía correctiva para resecar o crear los órganos reproductores apropiados para el sexo del niño. El tratamiento también puede incluir la terapia de reemplazo hormonal.
La correcta determinación del sexo es importante no sólo en lo que respecta al tratamiento sino también en lo relacionado con el bienestar emocional del niño. Estos pacientes deben ser criados en un ambiente donde se adapte con toda convicción al sexo «asignado».
Es probable que algunos de los niños que nacen con ambigüedad genital tengan órganos reproductores internos normales que les permitan llevar una vida fértil normal. Un tratamiento quirúrgico correcto y substitutivo hacen de ellos en la edad adulta, hombres viriles y mujeres capaces de relaciones sexuales satisfactorias. Sin embargo, otros niños pueden experimentar una reducción o la ausencia de fertilidad.
]]>Sexual differentiation disorders constitute a group of complex syndromes and entities. The term intersexual states makes reference to those newborns that present with ambiguos genitalia, that is, without any clear evidence of defined sex. Intersexuality may be classified as masculine and femenine pseudohermaphroditism and true hermaphroditism, which is caused by incomplete sexual differentiation in the male or virilization in the female. This disorder is relatively rare in our context, though knowledge increases more and more about it thanks to the development of molecular biology and the discovered involvement of genes in the normal sexual differentiation process. This article presented the case of a newborn whose dysmorphological exam at birth indicated positive findings in external genitalia such as micropenis and bifid scrotum. Labioscrotal fusion was upset with separated folds resembling labia majoris and a structure that seemed to be vaginal introitus. The ultrasonographic study eliminated the possibility of uterus and ovaria and showed the presence of testis in the inguinal canal. Sexual chromatin in interphase cells of the oral mucosa indicated 0% Barr corps and the chromosomal study showed normal male cariotype (46, XY), so this case was considered as male pseudohermaphroditism. Sexual differentiation disorders were reviewed.
Key words: sexual differentiation, disorders, ambiguos genitalia, pseudohermaphroditism, hermaphroditism, mixed gonadal dysgenesia.
1. Becker KL, Kenneth L. Sex determination and development. In: Principles and practice of endocrinology and Metabolism. Philadelphia: JB Lippincutt; 2003. Pp.788- 843.
2. Larsen WJ. Development of the urogenital system. In: Essentials of Human Embryology. Singapore: Churchill Livingstone; 2001. Pp.173-95.
3. Nussbaum RL, McInnes RR, Willard HF. Clinical Cytogenetics: Disorders of autosomes and Sex chromosomes. In: Thompson & Thompson Genetics in Medicine. 6th Ed. Philadelphia: WB Saunders; 2001. Pp. 157-65.
4. Villalobos P. Las claves. [en línea] 2002. Disponible en: http://www.el-mundo.es/cronica/2002/366/1035195281.html
5. Intersexualidad. [en línea] Revisado 2006. Disponible en: http://www.intersexualite.org/Spanish-Conway.html
6. González P, Quesada M, Cabrera R y Bello D. Disgenesia gonadal mixta con fórmula cromosómica 45,X /46,X, (mar). Presentación de una paciente. Rev Cubana Endocrinol. 2002; 13 (3). Disponible en: http://bvs.sld.cu/revistas/end/vol13_3_02/end07302.htm
7. La ambigϋedad genital. [en línea] Revisado 2006. Disponible en: http://www.baptisthealth.net/greystone/content.jsp?pageid=P06172
8. Genética, dos sexos, un cuerpo. En el pueblo de los hermafroditas. Crónica 2002. [en línea] Disponible en: http://www.el-mundo.es/cronica/2002/366/1035195281.html
9. Behrman RE, Kliegman RM. The urinary system and pediatric gynecology. In: Nelson essential of pediatrics. Philadelphia: WB Saunders; 2002. Pp.1332- 96.
10. Green M. Genitalia physical examination. In: Pediatric diagnosis. Philadelphia: WB Saunders; 2001. Pp.98- 103.
11. Hipogonadismo masculino. [en línea] Revisado 2006. Disponible en: http://www.medilegis.com/bancoconocimiento/H/Hipogonadismo/Hipogonadismo.asp
Recibido: 14 de octubre de 2006. Aprobado: 25 de enero de 2007.
Dra. Yoerquis Mejías Sánchez. Calle D. Betancourt y J. de la Vega, Camagüey, Cuba.
Correo electrónico: yoerme@finlay.cmw.sld.cu y yoerme@yahoo.es
1 Especialista de I Grado en Pediatría. Instructor del Hospital Pediátrico «Eduardo Agramante Peña» (Camagüey).
2 Especialista de II Grado en Epidemiología. Máster en Salud Ambiental.
3 Especialista de I Grado en Medicina General Integral. Especialista de I Grado en Genética Clínica. Instructor del Centro Provincial de Genética Médica, Villa Clara.