Revista Cubana de Pediatría. 2014; 86(1)
PRESENTACIÓN DE CASO
Gangliosidosis generalizada tipo 1
Generalized gangliosidosis type 1
]]>
Dr. Iván Hernández García,I Dra. Frances Seiglie Díaz,I Lic. Derbis Campos Hernández,II Lic. Lourdes Marrón Portarles,I Dr. Jorge Luís Díaz González,I Dr. Orlando Carmona PadrónI
IHospital Pediátrico Docente "William Soler". La Habana, Cuba.
IICentro Nacional de Genética Médica. La Habana, Cuba.
RESUMEN
La gangliosidosis generalizada tipo 1 es una enfermedad de acúmulo lisosomal producida por mutaciones en el gen de la enzima b-galactosidasa, caracterizada fundamentalmente por toma del sistema nervioso central, la visceromegalia, disostosis ósea y dimorfismo facial. Se presenta el caso de un lactante varón, hijo de padres no consanguíneos, de 5 meses de edad, Apgar 6/8 debido a hipoxia neonatal, con historia de múltiples ingresos por enfermedad diarreica e infecciones respiratorias. Es remitido a la Consulta de Genética Clínica por retardo del desarrollo psicomotor, macrocráneo y hepatomegalia, además de máculas hipercrómicas en piel. En el examen físico se encontraron evidencias de una posible afectación por enfermedad metabólica lisosomal. Entre las enfermedades a descartar estaban la galactosialidosis, de características clínicas similares, y la enfermedad de Morquio, con diferente presentación clínica pero idéntico defecto enzimático. ]]>
Palabras clave: gangliosidosis GM 1, enfermedad de acúmulo lisosomal, esfingolipidosis.ABSTRACT
Generalized or GM 1 gangliosidosis is a lysosomal storage disease caused by mutations in the enzyme b-galactosidase gene, mainly characterized by affecting the central nervous system, visceromegalia, osseous dysostosis and facial dimorphism. This is the case of a male nursling born to non-consanguineous parents, 5 months of age, Apgar index of 6/8 due to neonatal hypoxia, with a history of several admissions to hospital because of diarrheal disease and respiratory infections. He was referred to the clinical genetic service since he presented with retarded psychomotor development, macrocrania and hepatomegalia, in addition to hyperchromic skin spots. The physical exam found evidence of possible effects by lysosomal metabolic disease. Among the diseases to be ruled out for the diagnosis were galactosialidosis of similar clinical characteristics and Morquio B disease with different clinical presentation but identical enzymatic deficiency.
Keywords: GM 1 gangliosidosis, lysosomal storage disease, sphyngolipidosis.
INTRODUCCIÓN
La gangliosidosis generalizada tipo 1 (GM 1) es una enfermedad lisosomal, autosómica recesiva, producida por la deficiencia de la enzima b-galactosidasa ácida y la acumulación del gangliósido tipo 1 (GM 1), de oligosacáridos y de keratán sulfato (KS).1 Su incidencia varía entre diferentes regiones geográficas, desde 1:100 000 o 1:200 000 en EE. UU., hasta 1:3 700 en la isla mediterránea de Malta.2 La deficiencia de la hidrolasa lisosomal b-galactosidasa ácida, causa 2 enfermedades clínicamente distintivas: la GM 1 y la enfermedad de Morquio tipo B (mucopolisacáridos tipo IV-B) (MQB). Se ha reportado solapamiento entre las mutaciones que causan una u otra enfermedad.1,3,4 A su vez, la GM 1 tiene 3 formas clínicas bien definidas. De ellas, el tipo 1 o infantil, con inicio de manifestaciones clínicas en el período de la lactancia, combina hallazgos de neurolipidosis (neurodegeneración, mancha rojo cereza en el fondo de ojo), con las características de las mucopolisacaridosis (visceromegalia, disostosis múltiple y fascie tosca).
La b-galactosidasa ácida cataliza la hidrólisis de la galactosa, en posición b terminal, de los glicoconjugados GM 1, y genera un gangliósido tipo 2 (GM 2). También degrada otros glicoconjugados que tienen una galactosa en posición b, como el KS.1,4 La deficiencia enzimática produce acúmulo de glicoconjugados en los tejidos corporales y en la orina. Dentro de las células de los individuos enfermos se han detectado concentraciones elevadas de GM 1, de su derivado el asialo gangliósido GM 1 (GA 1), de oligosacáridos ricos en galactosa derivados de glicoproteínas y de KS.1 Las membranas, particularmente las de las neuronas, son ricas en gangliósidos. Su acumulación y la de sus derivados (GA 1), se cree que causan las manifestaciones neurológicas de la GM 1. ]]>
PRESENTACIÓN DEL CASO
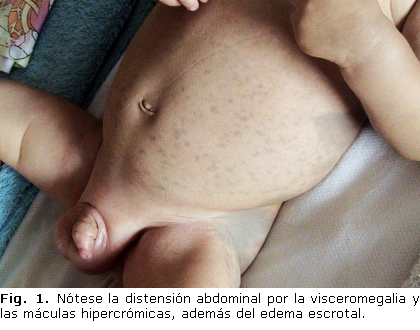
Lactante varón de 5 meses de edad, primer hijo de padres no consanguíneos, producto de parto eutócico, con Apgar 6/8 debido a hipoxia neonatal, con mensuraciones dentro de límites normales, e historia de múltiples ingresos por enfermedad diarreica aguda e infecciones respiratorias a repetición. Se remite a la Consulta de Genética Clínica por detectarse retardo del desarrollo psicomotor (no sostén cefálico, ni seguimiento visual), macrocráneo y hepatomegalia, además de máculas hipercrómicas en piel (figura 1).
Dismorfismo facial: cráneo de configuración normal y perímetro (45 cm) por encima del percentil 97, fontanela pequeña y abombamiento frontal. Se detectó desviación de hendiduras palpebrales hacia arriba, cejas pobladas, nariz bulbosa, edema periorbital, orejas rotadas posteriormente y labios gruesos. Se encontró asimetría de miembros inferiores: izquierdo más corto, con sindactilia 2/3 de ambos pies y edema escrotal (figura 1). Es llamativa la presencia de máculas melanodérmicas fundamentalmente en torso, más grandes y oscuras o confluentes en región sacrococígea y glúteos. A la palpación abdominal se encontró hígado que rebasa reborde costal.
Examen oftalmológico: nistagmo horizontal de gran amplitud y baja frecuencia con fase rápida a la derecha. El niño no sigue los objetos, solo la luz. El fondo de ojo arrojó disco pálido, atrófico, borde definido, capa de fibras ganglionares de aspecto amarillento con fóvea de color rojo preservado, y presencia de cambios pigmentarios de la mácula, propia de la distrofia de conos.
Diagnóstico presuntivo: enfermedad de almacenamiento lisosomal, esfingolipidosis, gangliosidosis GM 1. ]]>
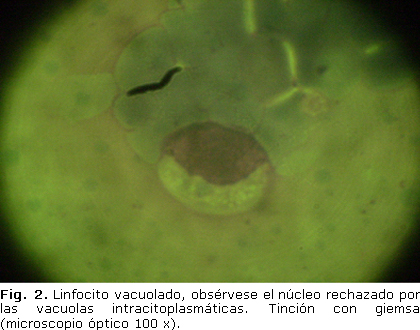
Exploración ósea: desproporción craneofacial, huesos wormianos, asimetría femoral (fémur izquierdo más pequeño, extremidad proximal y distal del fémur derecho amplia).El examen ultrasonográfico mostró hepatomegalia y la ecocardiografía fue normal. La tomografía simple de cráneo no reveló alteraciones. En la lámina periférica se observaron vacuolas intracitoplasmáticas de color blanco, con la tinción clásica de giemsa (figura 2).
Pruebas metabólicas: aminoaciduria inespecífica, aminoacidemia inespecífica y oligosacariduria. La cromatografía en placa delgada de mucopolisacáridos en orina fue normal. Las enzimas lisosomales arrojaron reducida actividad de b-galactosidasa ácida en leucocitos (0,18 nmol/mg proteína/hora), lo cual representó un 4 % de la actividad enzimática del control negativo.
DISCUSIÓN
El déficit de b-galactosidasa produce 2 enfermedades bien distintivas: la MQB y la GM 1, a su vez con 3 formas clínicas bien caracterizadas. La enfermedad de MQB es una entidad también lisosomal, con manifestaciones prácticamente privativas del sistema osteomioarticular.4,5 Las primeras manifestaciones de esta enfermedad aparecen entre el primer y tercer año de vida, y entre sus características distintivas se encuentran: la ausencia de síntomas y signos neuropatológicos, un neurodesarrollo normal y la presencia de KS en la orina. Aunque el paciente presenta disostosis ósea, dada por la discrepancia de los miembros inferiores, el inicio de su enfermedad fue más temprano, y predominaban las manifestaciones extraarticulares, características de la GM 1.
La galactosialidosis es una enfermedad de almacenamiento que comparte hallazgos clínicos tanto de las gangliosidosis como de las sialidosis. Se produce por un déficit combinado de la b-galactosidasa ácida y la neuraminidasa, secundario a un defecto de la L-carboxipeptidasa, proteína protectora que forma un complejo de alto peso molecular con las anteriores dentro del lisosoma.6 La actividad de ambas enzimas está reducida.
La galactosialidosis tiene 3 subtipos clínicos bien definidos: la forma infantil temprana, que se presenta en etapa prenatal, generalmente con hidropis fetal, o en promedio a los 14 días de nacido, con edema, ascitis y visceromegalia, a lo que se suma la displasia esquelética y la muerte temprana, cuadro sintomatológico diferente al de este paciente. La forma infantil tardía (16 meses), que se caracteriza por hepatoesplenomegalia, retardo del crecimiento, toma cardiaca, y por carecer de signos neurológicos relevantes, por lo que también se consideró conveniente descartarla. La mayoría de los casos reportados pertenecen al tipo juvenil o adulto (tercer subtipo), y son mayormente de origen japonés. El mioclono, la ataxia, el angioqueratoma, la discapacidad intelectual, la ausencia de visceromegalia y la supervivencia prolongada, son sus características distintivas.6 ]]>
El cuadro clínico y la determinación enzimática permitieron finalmente llegar al diagnóstico definitivo, y ofrecer un pronóstico a la familia del paciente. Además, permitirá el diagnóstico prenatal futuro, y brindar opciones reproductivas a la pareja, si esta decidiera tener nueva descendencia.
REFERENCIAS BIBLIOGRÁFICAS
1. Suzuki Y, Sakuraba H, Oshima A. b-Galactosidase deficiency (b-Galactosidosis): GM 1 Gangliosidosis and Morquio B Disease. En: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Online Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill [libro en Internet]; 2012 [citado 23 de enero de 2013].
Disponible en: http://www.ommbid.com/OMMBID/the_online_metabolic_and_molecular_bases_of_inherited_disease /b/abstract/part16/ch151
2. Tegay DH. GM1 Gangliosidosis. En: Medscape Week in Review [homepage en Internet] [citado 23 de enero de 2013]. Disponible en: https://login.medscape.com/login/sso /getlogin?urlCache=aHR0cDovL3d3dy5tZWRzY2FwZS5jb20vdmlld3Byb2dyYW0vNzA0OV9wbnQ=&ac=401
3. Caciotti A, Garman SC, Rivera-Colón Y, Procopio E, Catarzi S, Ferri L, et al. GM 1 gangliosidosis and Morquio B disease: an update on genetic alterations and clinical findings. Biochim Biophys Acta [serie en Internet]. 2011 Apr [citado 23 de enero de 2013];7. Disponible en: http://preview.ncbi.nlm.nih.gov/pubmed?term=generalizedgangliosidosis&cmd=correctspelling
4. Elizabeth F. Neufeld, Joseph Muenzer. The Mucopolysaccharidoses. En: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Online Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill [libro en Internet]; 2013 [citado 23 de enero de 2013]. Disponible en: http://www.ommbid.com/OMMBID/the_online_metabolic_and_molecular_bases_of_inherited_disease/b /abstract/part16/ch136
5. Chen H. Mucopolysaccharidosis IV (Morquio Syndrome). Atlas of Genetic Diagnosis and counseling. Totowa, New Jersey: Humana Press Inc.; 2006. p. 687-91.
6. Alessandra d'Azzo, Generoso Andria, Pietro Strisciuglio, Hans Galjaard. Galactosialidosis. En: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Online Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill [libro en Internet]; 2010 [citado 23 de enero de 2013]. Disponible en: http://www.ommbid.com/OMMBID/the_online_metabolic_and_molecular_bases_of_inherited_disease/b /abstract/part16/ch152
Recibido: 3 de mayo de 2013.
Aprobado: 9 de septiembre de 2013.
]]>
Iván Hernández García. Hospital Pediátrico Docente "William Soler". San Francisco # 10 112, Reparto Altahabana, municipio Boyeros. La Habana, Cuba.
Correo electrónico: ivan.hernandez@infomed.sld.cu ]]>