Diagnóstico y tratamiento de la enfermedad de Pompe
Diagnosis and treatment of Pompe disease
Norma Elena de León-Ojeda,I Frances Seiglie-Díaz,I Alina García-García,I Hiram Tápanes-Daumy,II Caridad Verdecia Cañizares,I Tatiana Acosta Sánchez,III Lázara Enma Larrinaga Vicente,III Cristina León DomínguezIII
IHospital Pediátrico "William Soler". La Habana, Cuba.
IICardiocentro Pediátrico "William Soler". La Habana, Cuba. ]]>
IIICentro Nacional de Genética Médica. La Habana, Cuba.
RESUMEN
Introducción: la enfermedad de Pompe es una glucogenosis tipo II con patrón de herencia autosómico recesivo, causada por la deficiencia de la enzima alfa-glucosidasa ácida, que provoca acúmulo de glucógeno en los lisosomas de los macrófagos. El gen está mapeado en el cromosoma 17q25, y han sido detectadas más de 460 mutaciones. Están descritas 3 formas clínicas de la enfermedad, que incluyen, la variedad infantil, la juvenil y la tardía. La variedad infantil se caracteriza por miocardiopatía hipertrófica o infiltrativa, hipotonía, hepatomegalia y macroglosia. En la variedad juvenil hay fiebre recurrente, infecciones respiratorias a repetición y discapacidad motora progresiva. La terapia de reemplazo enzimático modifica el curso clínico de la enfermedad.
Presentación de casos: se presentan los elementos al diagnóstico de 4 casos con esta enfermedad, 3 de ellos con la variedad infantil y uno con la variedad juvenil, y se discuten los resultados de estudio molecular. Se informa la evolución clínica de 2 de ellos, luego de 6 meses y un año de tratamiento con la terapia de reemplazo enzimático con alfa-glucosidasa ácida (myozyme), lo que ha llevado a la mejoría de parámetros bioquímicos y clínicos en ambos casos.
Conclusiones: en pacientes con enfermedad de Pompe la cuidadosa evaluación clínica para diagnosticar precozmente la enfermedad y el comienzo de la terapia de reemplazo enzimático contribuyen a mejorar las funciones musculares, motoras y cardiovasculares, lo cual impacta en la supervivencia y calidad de vida de estos niños.
Palabras clave: enfermedad de Pompe; enfermedades lisosomales; glucogenosis tipo II; terapia de reemplazo enzimático; alfa-glucosidasa ácida.
ABSTRACT
Introduction: Pompe disease is a type II glycogenosis with recessive autosomal inheritance pattern, which is caused by the acid alpha-glucosidase enzyme that brings about buildup of glycogen in macrophage lysosomes. The gene is already mapped in Chromosome 17q25 and has been so far detected over 460 mutations. Three clinical forms of disease were described, including infantile, juvenile and late-onset varieties. The infantile variety is characterized by hpertrophic or infiltratin myocardiopathy, hypotonia, hepatomegaly and macroglossia. The juvenile type comprises recurrent fever, repeated respiratory infections and progressive motor disability. Enzyme replacement therapy changes the clinical course of disease. ]]>
Case presentation: the elements for diagnosis of 4 cases were submitted, 3 of them with infantile form and one with juvenile one. The results of the molecular study were discussed. The clinical progress of 2 of them after 6 monts and one year of treatment with the enzymatic replacement therapy using acid alpha-glucosidase (myozyme), which led to improvement of biochemical and clinical parameters in both cases.
Conclusions: careful clinical assessment of patients with Pompe disease to early diagnose it and the onset of the enzyme replacement therapy help to improve muscle, motor and cardiovascular functions, which has an impact on survival and quality of life of these children.
Keywords: Pompe disease; lysosomal diseases; type II glycogenosis; enzyme replacement therapy; acid alpha-glucosidase.
INTRODUCCIÓN
La enfermedad de Pompe, también conocida como glucogenosis tipo II, es un error innato del metabolismo de los glúcidos causado por un déficit de alfa-glucosidasa ácida (GAA) o maltasa ácida. Tiene herencia autosómica recesiva y provoca acúmulo de glucógeno en los lisosomas en el músculo esquelético, cardiaco y también en otros órganos y sistemas que disfuncionan.1,2
Esta enfermedad tiene un espectro clínico amplio, que va desde el fenotipo clásico infantil, hasta las variedades moderadas que se presentan en niños y adultos, y tiene heterogeneidad clínica, pues una misma mutación puede tener variedad de formas de presentación en una misma familia. Se han identificado más de 460 mutaciones del gen que codifica esta enzima, ubicado en el cromosoma 17q25.2-4
Se reconocen dos formas de presentación en la infancia: la clásica, con una frecuencia estimada de 1:40 000; y la de inicio tardío, infraestimada en 1:57 000, aunque otros estudios reportan desde 1:14 000 a 1:300 000. La forma clásica es grave, evoluciona rápidamente a la disfunción cardiovascular, la hipotonía muscular, la visceromegalia y el deceso antes del año de vida. La forma de inicio tardío no es grave, ni de evolución tan rápida como la clásica. Aunque existen casos asintomáticos y su expresión clínica es variable, se caracteriza por debilidad muscular progresiva e insuficiencia respiratoria.4,5
Con el objetivo de evaluar el comportamiento de las variables clínicas, ecocardiográficas y de laboratorio de 4 pacientes en edad pediátrica con diagnóstico de enfermedad de Pompe, así como la evolución después del tratamiento sustitutivo enzimático en dos de ellos, es que se realiza este trabajo.
]]>PRESENTACIÓN DE CASOS
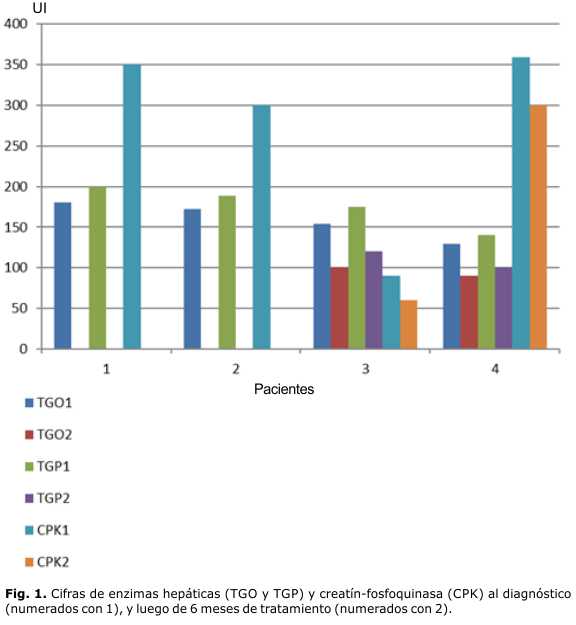
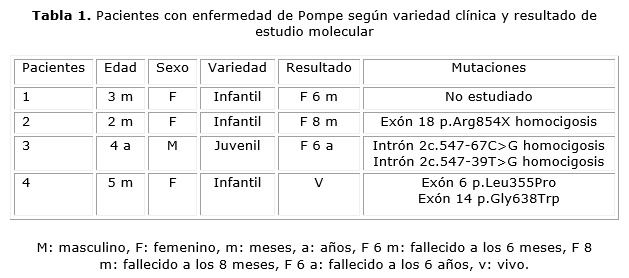
Fueron estudiados por sospecha clínica de la enfermedad 4 pacientes, 3 del sexo femenino con la variedad infantil. La forma de presentación en estas fue en los primeros 6 meses de vida, con hipotonía y reflejos osteotendinosos disminuidos, hepatomegalia, miocardiopatía hipertrófica de características infiltrativas, e infecciones respiratorias a repetición. Todas tuvieron cifras elevadas de transaminasas, creatin-fosfoquinasa (CPK) y la actividad enzimática en leucocitos fue baja. La macroglosia solo se presentó en las 2 pacientes afroamericanas, una de ellas no pudo ser estudiada molecularmente, y la otra resultó homocigótica para la mutación en el exón 18 c.2560C>T, que causa un cambio en la proteína p.Arg854X (tablas 1 y 2 y figura 1).
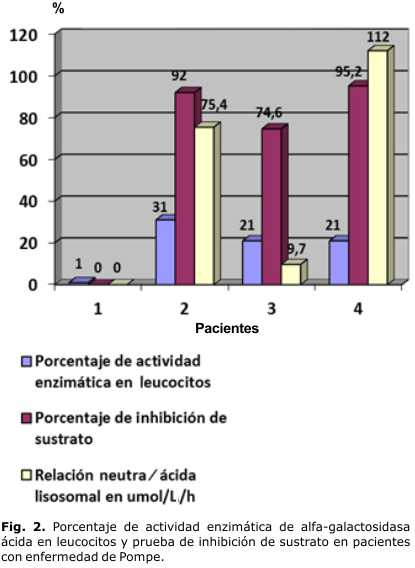
La paciente 4, con variedad infantil, comenzó con manifestaciones digestivas de intolerancia a los lácteos, y luego de los 4 meses fue apareciendo la hipotonía con reflejos osteotendinosos disminuidos, hepatomegalia, miocardiopatía y trastornos de deglución. Tuvo hipertransaminasemia, aumento de la CPK, oligosacariduria, actividad enzimática disminuida en leucocitos, y la inhibición de sustrato en rango de positividad. Se le tomó biopsia muscular con resultados no relevantes, y en el estudio molecular fue heterocigótica compuesta para dos mutaciones en este gen, una de ellas involucra el exón 6 c.1064T>C y el exón 14 c.1912G>T (tablas 1 y 2, y figuras 1 y 2).
El comienzo de la terapia de reemplazo enzimático (TRE) en esta paciente se efectuó desde los 8 meses con GAA (myozyme) a 20 mg por kg por dosis cada dos semanas. Requirió ingreso en unidad oncológica para acompañar la enzima con methotrexate a 0,4 mg∕kg∕dosis subcutáneo 15 min antes de la administración de la enzima, y continuar los dos días siguientes con igual dosis, durante las tres primeras administraciones. Este esquema de tratamiento también se acompañó de ondansetrón para minimizar las náuseas y regurgitaciones; no obstante, se presentaron complicaciones, como mucositis y eritema malar.
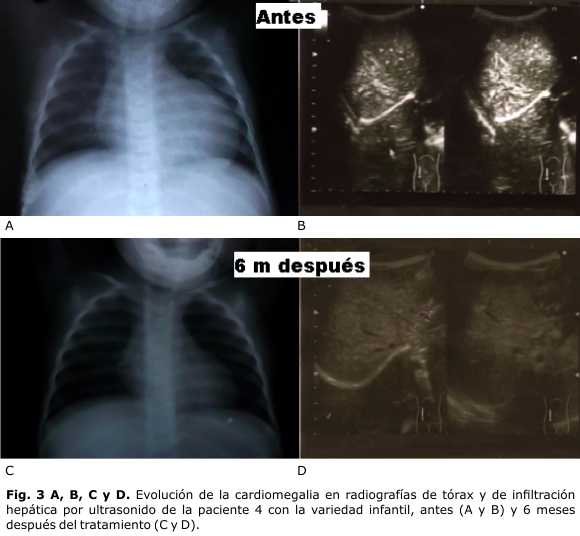
La función cardiovascular se encontraba severamente dañada en esta paciente con infiltración miocárdica concéntrica, que limitaba la cavidad del ventrículo izquierdo a un espacio virtual con fracción de acortamiento del 42 % y de eyección del 72 %. Luego de 6 meses de TRE ha mejorado la función cardiovascular hasta la disminución de la infiltración miocárdica y medidas del tabique interventricular de 7 mm, con fracciones de eyección de 75 % y de acortamiento del 41 %, ondas E 1,05 y A 35,7, y relación E∕A de 2,06. El ecocardiograma evolutivo muestra una miocardiopatía infiltrativa en fase de resolución. La cardiomegalia, que era ostensible al comienzo de la TRE, remitió en las radiografías a los 6 meses de comenzada la terapia. La infiltración de glucógeno hepático visible por ultrasonido, fue disminuyendo al mes de tratamiento hasta desaparecer luego de la tercera dosis; y en cuanto a la hepatomegalia, que era de 4 cm al comienzo, hubo disminución hasta desaparecer al día 70, luego de la quinta dosis (figura 3).
En cuanto a las funciones neurológicas ha habido una ganancia de habilidades motoras. Desde el sostén cefálico adecuado a los 14 días después de la primera administración, luego del mes de terapia, se volteaba prono-supino y supino-prono, ya sin ayuda de la cabeza como impulsora del movimiento. A los dos meses llevaba objetos a la línea media y comenzó a reptar, aún sin lograr apoyo en 4 puntos; además, se sostenía sentada, y se incorporaba con ayuda a esta posición, ya con sostén de tronco. A los 84 días se sentaba sola, y movía el tronco con mejoría de la motricidad fina. A los 6 meses, luego del comienzo de la TRE, ya dio sus primeros pasos con apoyo.
]]> Desde el punto de vista nutricional, se alimenta con licuado hiperproteico y leche de soja, pero el aumento de peso con ganancia de área grasa y muscular fue visible en la antropometría, a medida que se avanzó con la sustitución enzimática.El paciente 3, con la variedad juvenil, inició la enfermedad luego del año de vida, con discapacidad progresiva para la marcha, dada por debilidad de la cintura pélvica y escapular, con reflejos osteotendinosos disminuidos. Comenzó a presentar cambios posturales para la bipedestación, con rectificación de la lordosis lumbar, contractura y acortamiento de la musculatura isquiotibial de ambos miembros inferiores, y anadeo, por la debilidad de la cintura pélvica y de los músculos paravertebrales lumbosacros, que recordaban la postura de la espina rígida. También presentaba fiebre intercurrente, con infecciones respiratorias recurrentes, hepatomegalia y diarreas a repetición, que llevaron al diagnóstico de un quiste de colédoco a los 3 años, por lo cual se practicó colecistectomía con derivación yeyuno-ileal (tabla 2). A los 4 años, durante ingreso para estudio en esta institución por fiebre de causa desconocida, se le realizaron estudios químicos y sanguíneos que mostraron hipertransaminemia, aumento de la lactato-deshidrogenasa (LDH) y CPK normal. En el ultrasonido abdominal se constató hepatomegalia, ecocardiografía con morfología de la emergencia aórtica ectásica a nivel de anillo, bulbo y unión sinotubular, y en la electromiografía y potenciales somatosensoriales, apareció daño miopático, y a la vez, neuropático (figura 1).
Fue interconsultado con Genética Clínica, y se le realizó estudio de orina, en el cual se constató presencia de oligosacáridos por cromatografía de capa delgada, y se procedió a la cuantificación de la enzima alfa-glucosidasa ácida en leucocitos, con valor de 3,53 nmol∕mg∕h para 22,2 % de actividad respecto al control. La reacción ácida-neutra lisosomal fue de 9,7 µmol/L/h (normal) y la inhibición de sustrato fue de 74,6 %, negativa según valores de referencia, pero complementaria con la actividad enzimática en leucocitos (figura 2).
En este caso el resultado del estudio molecular en sangre reveló la existencia de dos mutaciones en condición homocigótica en el gen GAA, c.547-67C>G y c.547-39T>G, ambas en el intrón 2 del cromosoma 17q25. Tuvo además 120 variantes polimórficas conocidas (tabla 1).
En cuanto a la realización de otras pruebas como las funcionales respiratorias, no hubo cooperación por la edad del paciente al diagnóstico. No pudo enviarse muestra para estudio de CRIM (Cross-Reactive Immunological Material) ni presencia de anticuerpos contra la enzima. Se comenzó con la TRE hace ya un año con GAA, con administración cada dos semanas a 20 mg∕kg por dosis endovenosa con bomba de infusión, cambios en el ritmo de la administración cada 30 min, y monitoreo de la función cardiovascular y respiratoria. En este tiempo no se presentaron reacciones adversas, y los parámetros hemoquímicos mostraron discreta mejoría en las cifras de transaminasas (figura 1).
Relativo a las funciones motoras, la prueba de la marcha de 6 min no era realizable en este paciente, pues al comienzo necesitaba apoyo, y un año después logró liberarse del apoyo, pero continuó con debilidad de miembros inferiores y debilidad muscular de paravertebrales.
Luego de cuadro diarreico crónico y pancreatitis aguda, este paciente, con 6 años de edad, hizo éxitus por coagulación intravascular diseminada. Las cifras de IgM y complemento fueron normales, y la tinción de ácido periódico de Schiff (PAS) no mostró acúmulo de glucógeno en el hígado ni el miocardio.
DISCUSIÓN
Los 4 pacientes tuvieron manifestaciones clínicas clásicamente descritas de esta enfermedad en sus respectivas variantes. Todos tuvieron excreción de oligosacáridos en orina, hipertransaminasemia, y los 3 con variedad infantil mostraron aumento de la CPK. En cuanto a la prueba de inhibición de sustrato, fue positiva en los casos estudiados, excepto en el paciente de la variedad juvenil, que sí fue proporcional a la actividad enzimática en leucocitos.
]]> En el estudio molecular la mutación de la paciente 2 en el exón 18 c.2560C>T, causa un cambio en la proteína p.Arg854X, y ha sido descrita en afroamericanos con frecuencia de 5 a 7 %, presentación muy severa, rápida progresión de las manifestaciones clínicas y letalidad precoz, como ocurrió en este caso, que falleció a los 6 meses.1,6,7La paciente 4 fue heterocigótica compuesta para dos mutaciones en este gen, una en el exón 6 c.1064T>C, que causa cambios proteicos p.Leu355Pro y en el exón 14 c.1912G>T, que conlleva a la sustitución en la proteína p.Gly638Trp. Estas mutaciones son consideradas potencialmente menos severas, pero en este caso, pudieran justificar la aparición de las manifestaciones clínicas más tardíamente y su progresión menos rápida, lo cual permitió confirmar el diagnóstico y comenzar el tratamiento sustitutivo a los 8 meses en que primaba la hipotonía global con retraso del neurodesarrollo progresivo, y solo quedaba sostén cefálico pobre con movimientos voluntarios de los segmentos distales de los miembros.6
Las manifestaciones más frecuentes en la variedad infantil incluyen la cardiomegalia, la dificultad respiratoria, la debilidad muscular, los trastornos de deglución y el retraso del desarrollo, como ocurrió en esta paciente.7,8
La infiltración miocárdica concéntrica afecta la fracción de eyección de ventrículo izquierdo, que fluctúa en un rango de 18-73 %. Algunos pacientes que comenzaron tratamiento entre 6 y 7 meses, tuvieron fracciones de eyección iniciales de 18 % y a los dos meses de tratamiento llegaron a 25 y 65 %, aunque fueron medidas por resonancia magnética. En este caso hubo infiltración miocárdica con tabique de 15 mm y fracción de eyección de 10 %, que se ha incrementado a 84 % luego de 6 meses de tratamiento, y el tabique ha disminuido a 8 mm. Las mediciones fueron realizadas por ecocardiografía y sin sedación.7-9
En cuanto a la infiltración de glucógeno hepático, tanto en la variedad infantil como en la juvenil, se reporta la hepatomegalia, que mejora luego del tratamiento, como sucedió en ambos pacientes que recibieron TRE.1
El paciente 3, con la variedad de inicio tardío, comenzó con discapacidad para la marcha después del año de vida, y se describe que el 67 % de los pacientes con esta variedad sufre de debilidad y calambres en los miembros inferiores, con disminución progresiva de la fuerza muscular, y cerca del 20 % de los niños afectados presenta algún síntoma antes de los 12 años. Las infecciones respiratorias a repetición, la fiebre intercurrente, la hepatomegalia y la discapacidad motora, fueron los síntomas que acompañaron el diagnóstico del paciente con la variedad juvenil, como también se describe en la literatura.5,6 Este caso tuvo una reacción ácida-neutra lisosomal de 9,7 umol/L/h (normal) y la inhibición de sustrato fue de 74,6 %, negativa según valores de referencia, pero complementaria con la actividad enzimática en leucocitos. El uso de sangre seca DBS (dried blood spots) para diagnóstico, tiene ventajas para pesquisa por la estabilidad de la técnica, sin embargo, la confirmación del diagnóstico se realiza por medio de cuantificación enzimática en leucocitos (que estuvo por debajo del 30 % respecto al control en este caso), o estudio molecular.7,10
A pesar de que en el intrón 2 no se han descrito eventos relacionados con el empalme como en los intrones 1, 10 o 17, sí se ha relacionado otro caso con clínica y mutaciones idénticas a las reportadas por Guevara-Campos en un paciente venezolano con enfermedad de Pompe variante juvenil, que tampoco mostraba una disminución significativa de su actividad enzimática en papel de filtro, como ocurrió en este caso.6,11
Llama la atención que estos 2 pacientes, que tienen una clínica similar con CK normal, pertenecen a grupos poblacionales diferentes, y tienen exactamente las mismas variantes polimórficas, con idénticos cambios en los mismos intrones y exones. Se ha descrito la presencia de efectos fenotípicos al confluir variantes polimórficas en otras enfermedades lisosomales, como en la enfermedad de Fabry, lo cual no se refiere en la de Pompe.12
La electromiografía mostró signos de daño miopático y neuropático. En otros casos se reportan patrones repetitivos complejos o miotónicos, sobre todo, en la musculatura paraespinal.13 En animales transgénicos modelos para esta enfermedad, se ha estudiado el comportamiento de la unión neuromuscular y se comprobaron cambios neuropáticos possinápticos y presinápticos, como la reducción de los niveles de proteínas en neurofilamentos, la disminución del diámetro axonal y del grosor de la mielina, lo cual corrobora la pérdida de integridad de la unión neuromuscular.14
La presencia de infiltración en el sistema nervioso central se reporta en casos con enfermedades lisosomales, sobre todo, en el tálamo, la corteza, el hipocampo y el cerebelo, pero es menos frecuente en la enfermedad de Pompe. Se han descrito casos con acúmulo de glucógeno cortical, periventricular, en las astas anteriores de la médula espinal y cambios en la sustancia blanca. La atrofia muscular se relaciona con el acúmulo de glucógeno, pero también con la disfunción mitocondrial, la autofagia y la apoptosis, y es más discapacitante cuando toma la musculatura paravertebral.15,16
]]> En cuanto a herramientas para la evaluación motora, en la mayor parte de las publicaciones se habla de la prueba de la marcha de los 6 min que a este paciente le resultaba imposible realizar; recientemente se ha sugerido que no se tome como referente en la evaluación de los casos.5 La escala del Medical Research Council, que se usa para medir la fuerza de los grupos musculares deseados, no fue aplicada en este paciente, pues ha sido recientemente validada y sugerida para su seguimiento.17En cuanto a las escalas funcionales compuestas, la más relevante es la North Star Ambulatory Assessment, diseñada específicamente para niños con distrofia muscular de Duchenne en su etapa ambulante, pero aún no se ha demostrado su validez en niños con enfermedad de Pompe.18,19
La escala Raschbuilt Pompe-specific Activity (R-PAct), o escala de actividad específica de la enfermedad de Pompe, que detecta limitaciones en las actividades y en la participación social a lo largo de todo el espectro de la enfermedad, ha sido recomendada recientemente por comité de expertos, por lo que no fue aplicada en este paciente.5,20
Otro síntoma frecuente e incapacitante en la enfermedad de Pompe es la fatiga, cuya intensidad debe medirse por escala validada en español, que incluye una serie de preguntas al paciente cuyas respuestas se puntúan del 1 al 7, a pesar de que las respuestas son subjetivas, y mucho más a esta edad, por lo que tampoco fue aplicada.21
Se han hecho estudios para diagnóstico de esta entidad en pacientes con patrón de debilidad de cintura escapular y miembros, síntomas respiratorios, escoliosis o espina rígida y diagnóstico no conocido. De 102 casos estudiados, hubo 6 positivos.22
De 4 pacientes diagnosticados en la India con la variante juvenil, se comenzó tratamiento en 2 de ellos a los 8 y 14 meses, y ambos fallecieron con 14 y 15 meses por insuficiencia respiratoria.23
En 22 estudios de 437 pacientes en total con TRE, al menos 2 tercios de ellos se estabilizaron o mejoraron los niveles de CPK, y la función muscular o respiratoria. Las reacciones adversas fueron ligeras o moderadas, y la enzima atenúa la progresión de la enfermedad en la mayoría de los pacientes.24
El 40 % de los pacientes desarrollan respuesta inmunológica a la terapia. La expresión endógena de GAA se determina por el CRIM. En un estudio de 11 pacientes, todos desarrollaron anticuerpos, los 3 CRIM negativos tuvieron peor pronóstico y mejoró poco la función cardiovascular.25,26
En estos casos no pudo realizarse la toma de muestras para el estudio de CRIM ni se cuantificaron los niveles de anticuerpos contra la enzima. En relación con el tratamiento de la enfermedad con TRE, hay acuerdo en que es eficaz en lograr la mejoría y la estabilización de la función motora y pulmonar, como ha sucedido clínicamente con estos pacientes.5 Luego de la TRE la paciente 4, con la variedad infantil, ha mostrado signos de mejoría en su desarrollo psicomotor, con ganancia progresiva de habilidades motoras, pues ha logrado sentarse, caminar y movimientos del tronco en menos de un año de sustitución enzimática, además de la mejoría cardiovascular y la disminución del acúmulo de glucógeno hepático.
En el paciente 3, con la variedad juvenil, los progresos motores fueron más limitados y menos cuantificables, por ausencia de reglas de oro para demostrar la mejoría. Mantuvo dificultad para la marcha luego de un año de TRE; no obstante, mostró distanciamiento en cuanto a las infecciones respiratorias, disminución de la hipertransaminasemia y de la ecogenicidad hepática.
]]> Se concluye que aunque hubo casos en que los exámenes no fueron positivos para diagnóstico, los hallazgos clínicos son altamente sugestivos de la enfermedad. La aplicación del método clínico, en conjunción con los hallazgos bioquímicos, resulta indispensable para el diagnóstico de esta enfermedad genética. La TRE resulta beneficiosa para revertir los signos de acúmulo de glucógeno en estos pacientes.
Agradecimientos
Nuestro especial agradecimiento a Genzyme Sanofi Company por el apoyo incondicional que nos ha brindado en el diagnóstico y tratamiento de estos pacientes. A Marina Szlago y Joao Pesquero, por la inmediatez de sus resultados.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses en la realización del estudio.
REFERENCIAS BIBLIOGRÁFICAS
1. Lim JA, Li L, Raben N. Pompe disease: from pathophysiology to therapy and back again. Frontiers in Aging Neuroscience. 2014;6(177):1-14.
]]>2. Bergsma AJ, Kross M, Hoogeveen-Westerveld M, Halley D, van der Ploeg AT, Pijnappel P. Identification and Characterization of Aberrant GAA pre-mRNA Splicing in Pompe Disease Using a Generic Approach. Hum Mutat. 2015;36(1):57-68.
3. De Filippi P, Saeidi K, Ravaglia S, Dardis A, Angelini C, Mongini T, et al; and The Italian GSDII Group. Genotype-phenotype correlation in Pompe disease, a step forward. Orphanet Journal of Rare Diseases. 2014;9:102-13.
4. van der Meijden JC, Güngör D, Kruijshaar ME, Muir ADJ, Broekgaarden HA, van der Ploeg AT. Ten years of the international Pompe survey: patient reported outcomes as a reliable tool for studying treated and untreated children and adults with non-classic Pompe disease. J Inherit Metab Dis. 2015;38(3):495-503.
5. Gutiérrez-Rivas E, Illa I, Pascual-Pascual SI, Pérez-López J, Vílchez-Padilla JJ, Bautista-Lorite J, et al. Guía para el seguimiento de la enfermedad de Pompe de inicio tardío. Rev Neurol. 2015;60:321-8.
6. Pompe Center. Molecular aspects: mutations [homepage en Internet]; 2015 [citado 10 de julio de 2015]. Disponible en: http://cluster15.erasmusmc.nl/klgn/pompe/mutations.html
7. Diagnóstico y tratamiento de la Glucogenosis tipo II (enfermedad de Pompe) [homepage en Internet]; México: Secretaría de salud, 2010 [citado 10 de julio de 2015]. Disponible en: http://www.cenetec.salud.gob.mx/descargas/gpc/CatalogoMaestro/506_GPC_EnfPompe/GER_GlucogenosisII_Pompe.pdf
8. Wang RY, Bodamer OA, Watson MS, Wilcox WR, ACMG Work Group on Diagnostic Confirmation of Lysosomal Storage Diseases. Lysosomal storage diseases: diagnostic confirmation and management of presymptomatic individuals. Genet Med. 2011;13(5):457-84.
9. Barker P, Campbell MJ, Stephen D, Li J, Kim R, Pasquali S, et al. MRI assessment of left ventricular structure and function in children with infantile Pompe disease. J Cardiovasc Magn Resonance. 2010;12(suppl 1):309-10.
10. Horvath G, Sirrs S, Stockler S, Salvarinova-Zivkovic R, Vallance H, Waters P. Three years experience with dried blood spot a-glucosidase screening for Pompe disease in British Columbia, Canada. BMC Musculoskeletal Disorders. 2013;14(suppl 2):2.
11. Guevara-Campos J, Romeo-Villarroel MA, González-De Guevara L, Escobar V. Dos nuevas mutaciones en el gen que codifica la alfa-glucosidasa ácida en un adolescente con enfermedad de Pompe de inicio tardío. Rev Neurol. 2013;57(6):265-6.
12. Pisan A, Imbriaco M, Zizzo C, Albeggiani G, Colomba P, Alessandro R, et al. A classical phenotype of Anderson-Fabry disease in a female pa:ent with intronic mutations of the GLA gene: a case report. BMC Cardiovasc Disord. 2012;12:39.
]]>13. Park JS, Kim HG, Shin JH, Choi YC, Kim DS. Effect of enzyme replacement therapy in late onset Pompe disease: open pilot study of 48 weeks follow-up. Neurol Sci. 2015;36(4):599-605.
14. Falk DJ, Todd AG, Lee S, Soustek MS, ElMallah MK, Fuller DD, et al. Peripheral nerve and neuromuscular junction pathology in Pompe disease. Hum Mol Genet. 2015;24(3):625-36.
15. Platt FM, Boland B, van der Spoel AC. Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J Cell Biol. 2012;199(5):723-34.
16. Kishnani PS, Beckemeyer AA, Mendelsohn NJ. The new era of Pompe disease: Advances in the detection, understanding of the phenotypic spectrum, pathophysiology, and management. Am J Med Genet Part C Semin Med Genet. 2012;160C(1):1-7.
17. Wokke JHJ, Escolar DM, Pestronk A, Jaffe KM, Carter GT, Van den Berg LH, et al. Clinical features of late-onset Pompe disease: a prospective cohort study. Muscle Nerve. 2008;38:1236-45.
]]>18. Mazzone ES, Messina S, Vasco G, Main M, Eagle M, D'Amico A, et al. Reliability of the North Star ambulatory assessment in a multicentric setting. Neuromuscul Disord. 2009;19:458-61.
19. Pane M, Mazzone ES, Sivo S, Sormani MP, Messina S, D'Amico A, et al. Long term natural history data in ambulant boys with Duchenne muscular dystrophy: 36-month changes. PLoS One. 2014;9:e108205.
20. Van der Beek NA, Hagemans ML, Van der Ploeg AT, Van Doorn PA, Merkies IS. The Rasch-built Pompe-specific Activity (R-PAct) scale. Neuromuscul Disord. 2013;23:256-64.
21. Hernández-Vidal P, Berrios GE, Bulbena A. Concepto y evaluación de la sensación de fatiga. En: Bulbena A, Berrios GE, Fernández de Larrinoa P, eds. Medición clínica en Psiquiatría y Psicología. Barcelona: Masson. 2000. p. 123-35.
22. Willis T, Roberts M, Hilton-Jones D, Quinlivan R, Hanna M, Straub W. Detection rate of Pompe disease in undiagnosed neuromuscular patients from four major centres in the UK-results of a 12 month prospective audit. BMC Musculoskeletal Disorders. 2013;14(suppl 2):20.
]]>23. Lingappa L, Devi RR, Dherai A, Rehder C, Kishnani P. Pompe disease-experience from South India. BMC Musculoskeletal Disorders. 2013;14(suppl 2):23.
24. Schoser J. Alglucosidase alfa: 5 years of experience in late-onset Pompe disease. BMC Musculoskeletal Disorders. 2013;14(suppl 2):O9.
25. Austin S, Kishnani P. Long term outcome and clinical experience on immune tolerance induction therapies in infantile Pompe disease. BMC Musculoskeletal Disorders. 2013;14(suppl 2):O8.
26. van Gelder C, Kroos M, Özkan L, Plug I, Reuser A, van der Ploeg A. Antibody formation to enzyme therapy in classic infantile Pompe disease: implications of patient age. BMC Musculoskeletal Disorders. 2013;14(suppl 2):18.
]]> Recibido: 20 de agosto de 2015.
Norma Elena de León-Ojeda. Hospital Pediátrico Universitario "William Soler". San Francisco # 10 112, reparto Altahabana, municipio Boyeros. La Habana, Cuba. Correo electrónico: norma.deleon@infomed.sld.cu
]]>
{kind=link}
{kind=link}