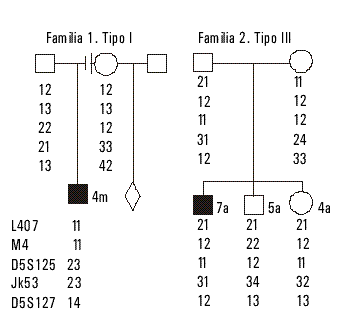
Fig. 1. Árboles genealógicos y haplotipos de las familias 1 y 2 descritas en el texto (ver resultados).
Resumen: La atrofia muscular espinal (AME) es un trastorno neurodegenerativo hereditario causado por la afectación selectiva de las motoneuronas del asta anterior de la médula espinal. Las atrofias musculares espinales son un importante grupo de enfermedades paralizantes que pueden afectar a los individuos de todas las edades y sexo. Forman un amplio espectro clínico genético que las distingue en 3 grupos de acuerdo con la edad de comienzo de los signos clínicos y la severidad de la enfermedad. Las 3 formas de AME autosómica recesiva fueron mapeadas en la región 5q 11.2-13.3, región que contiene múltiples copias de genes marcadores. Muchos fueron los estudios genéticos moleculares realizados para aislar el gen o genes que provocan la enfermedad, entre ellos, el análisis de ligamiento con marcadores polimórficos de la región, han permitido el estudio en familias afectadas. Precisamente este trabajo tuvo el objetivo de introducir en Cuba el diagnóstico molecular de la AME, para lo cual se evaluaron los marcadores del locus de la enfermedad: L407, M4, D5S125, D5S112, D5S127, D5S610, D5S1416, D5S107, D5S1356 y D5S557. Para el análisis de segregación alética se aplica la técnica molecular de reacción en cadena de la polimerasa (PCR), que permite identificar los cromosomas afectados a través de la obtención del genotipo en las familias diagnosticadas con los tipos clásicos I, II, III. En una familia donde el primero de 3 hijos es enfermo con el tipo III, se determinó que su hermanita de 4 años es portadora y su hermanito de 5 años es sano. Se realizó consejo genético a la familia.
Descriptores DeCS: ENFERMEDAD DE WERDNIG-HOFFMANN/genética; ENFERMEDAD DE WERDNIG-HOFFMANN/diagnóstico; CUBA.
Dentro de las enfermedades neuromusculares, los trastornos en el nivel medular suelen desarrollar un cuadro clínico donde los elementos predominantes son: debilidad muscular, atrofias musculares por denervación, disminución o pérdida de los reflejos musculares, hipotonía y en muchos casos fasciculaciones de los músculos de la lengua.1 En este grupo las AMEs son trastornos neurodegenerativos de causa genética hereditaria, producidos por la afectación selectiva de las neuronas motoras del asta anterior de la médula espinal, que pueden aparecer en edades muy tempranas de la vida y resultar fatales.
La atrofia muscular espinal de la infancia (AMEI) es considerada la segunda enfermedad autosómica recesiva fatal después de la fibrosis quística,2 con una incidencia estimada de 1/10 000 nacidos vivos, con una frecuencia de portadores3 que oscila para el tipo I entre 1/40 a 1/60. La heterogeneidad clínica de la AMEI fue reconocida lo que hizo que las interpretaciones de los resultados llevaran a varias clasificaciones. La considerable variación en la edad de comienzo y el curso clínico en diferentes pacientes, permitió la clasificación en 3 tipos de AMEI.4
La forma infantil más severa, tipo I, o enfermedad de Werdnig-Hoffmann, presenta un curso clínico más o menos homogéneo, que puede comenzar con manifestaciones antes del nacimiento o antes de los 6 primeros meses de vida, con deterioro progresivo que culmina con la muerte entre el primer y segundo años de vida.5 El tipo II, forma intermedia, con un comienzo antes de los 2 años y el tipo III o enfermedad de Kugelber-Welander, son formas clínicamente heterogéneas.6,7 En esta categoría no se incluyen los casos que comienzan entre los 17 y 55 años, llamados atrofias musculares del adulto y aquéllas con un modo de herencia ligada al cromosoma X llamadas enfermedad de Kennedy.8 Las 3 formas fueron localizadas9-12 en el mismo locus en el cromosoma 5q11.3-13.3.
Numerosos han sido los estudios genéticos moleculares realizados para localizar el gen o genes responsables de la AME, dentro de ellos el estudio de ligamiento con marcadores microsatélites polimórficos constituye un método empleado en el consejo genético en familias con AME autosómica recesiva. Muchas de estas familias deseaban tener un diagnóstico prenatal en el siguiente embarazo, que no era posible hasta el momento por no contar con alguna prueba útil para el diagnóstico. Ahora con el empleo de marcadores genéticos de la región del cromosoma 5q es posible brindar un diagnóstico a familias con al menos un niño previo afectado.
| Marcadores | Locus | No. de alelos | Tamaño (pb) | PIC reportados | PIC obtenido |
| 38,3 | | | | | |
| 2ae 9.1 | | | | | ]]> 0,50 |
| ET (TG/AG)n | | | | | |
| YN (CT)n | | | | | ]]> 0,93 |
| Afm 114ye7 | | | | | |
| 116-CA1 | | | | | ]]> 0,90 |
| 236B1-CA1 | | | | | |
| JK53-CA? | | | | | ]]> 0,83 |
| Mfa27 | | | | | |
Origen: Referencias 10,14,15,16 y 17. Resultados de la investigación.
Tabla 2. Condiciones de reacción de amplificación (PCR) para los marcadores utilizados en el estudio
]]>
| Locus | Temp. | MgC12 | Tiempo de |
| annealing | (mmol/L) | corrida (min) | |
| (° C) | |||
| D5S610 | | | |
| D5S557 | | | |
| D5S125 | | | ]]> 120 |
| D5S465 | | | |
| D5S127 | | | |
| D5S1416 | | | |
| D5S1356 | ]]> 60 | | |
| D5S112 | | | |
| D5S107 | | | |
Fig. 1. Árboles genealógicos y haplotipos de las familias 1 y 2 descritas en el texto (ver resultados).
El propósito presentó signos clínicos comparables con la AME tipo III, debilidad muscular y dificultades para caminar alrededor de los 7 años. La biopsia y la electromiografía fueron neurogénicas. Dos hermanos fueron también genotipados al tener riesgo de portar la enfermedad y pudo determinarse que el hermano de 5 años es sano y la hermanita de 4 años es portadora del cromosoma materno afectado.
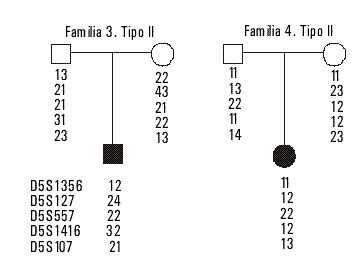
Fig. 2. Árboles genealógicos de las familias 3 y 4 descritas en el texto (ver resultados).
El caso índice en la familia 3 evaluado a los 2 años, presentó signos de hipotonía, debilidad para caminar comparable con la AME tipo II, al igual que el caso de la familia 4 diagnosticado a los 10 meses. Estas familias tienen solo un hijo y enfermo por lo que fue realizado el consejo genético.
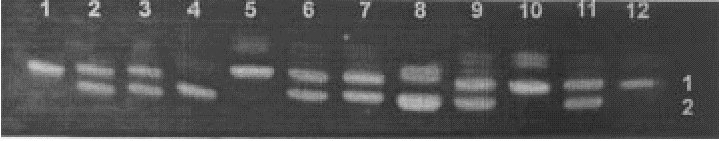
Fig. 3. Análisis de segregación en 4 familias afectadas con el marcador microsatélite D5S557 (ver texto en resultados).
De izquierda a derecha las 4 primeras carrileras corresponden a una familia donde el padre (1) y la madre (4) son homocigóticos para el alelo 1 y 2 respectivamente. El caso índice (2) y el (3), su hermano, aparentemente sano, son heterocigóticos. De 5 a 8 es otra familia que presenta un patrón de bandas similar al obtenido en la familia 1. Por el resultado que se muestra en estas familias este marcador es poco informativo ya que los padres son homocigóticos y los hermanos tienen la misma herencia alélica, pero no se pudo determinar cuál es el alelo afectado. En la tercera familia, carrileras de la 9 a la 12, resultó un patrón donde el padre (9) es heterocigótico, la madre (12) y el niño enfermo (10) son homocigóticos para el alelo 1, el segundo hijo (11) es heterocigótico y portador de la mutación. En esta familia el alelo 1 es el afectado.
Muchas familias portadoras de AME deseaban el estudio genético en un nuevo embarazo, que sólo se realizaba a través del consejo genético donde el riesgo de recurrencia es de 1 en 4, es decir, 25 % en cada embarazo por ser una enfermedad autosómica recesiva. Con el empleo del análisis de ligamiento utilizando marcadores polimórficos de la región del gen de la AME se brinda la posibilidad de realizar el diagnóstico molecular en las familias afectadas.
Esta experiencia en la aplicación del diagnóstico molecular de las formas de AME es una clara ventaja en la complementación del estudio genético molecular de la enfermedad.
Sin embargo, la exactitud del diagnóstico prenatal depende de la distancia genética entre los marcadores y el locus de la enfermedad y de la estructura de la familia, que hace necesario contar con una serie de marcadores óptimos cercanos al locus, teniendo en cuenta su informatividad dentro de cada familia. De esta forma es importante seguir un criterio diagnóstico estricto, basado en los estudios clínicos, electromiografías y biopsias de músculo positivas antes de realizar el consejo genético. ]]>
Cuando en las familias portadoras el niño ha fallecido, no existe sangre, músculo u otros tejidos que permitan la obtención de ADN, el estudio indirecto deja de ser útil, por ello es de gran importancia la captación de las familias con niños que presentan signos de la enfermedad desde los primeros meses de vida, fundamentalmente en los tipo I, donde la espectativa de vida es tan reducida. De esta forma resulta importante determinar el genotipo en la familia utilizando marcadores moleculares. Sin embargo, la predicción de un feto enfermo utilizando el método directo de identificación de los genes candidatos19?21 resulta un diagnóstico más rápido y exacto para el diagnóstico y brinda la posibilidad de realizar un consejo genético adecuado.Summary: The spinal muscular atrophy (SMA) is a hereditary neurodegenerative disorder caused by selective affection of motor neurones in the anterior of the spinal cord. The spinal muscular atrophics are an important group of paralyzing diseases that may affect any individual regardless of age or sex and they make up a wide clinical genetic range classified into 3 groups interms of age of onset of clinical signs and disease severity. The 3 forms of recessive autosomal SMA were mapped in 5q 11.2-13.3 region which contains several copies of marker genes. Many molecular genetic studies such as linkage analysis with polymorphic markers of the region carried out to isolate that gene or those genes causing the disease have allowed to study the affected families. Hence this paper is aimed at introducing the SMA molecular diagnosis in Cuba for which disease locus markers are evaluated i.e, L407;M4, D5S125, D5S127, D5S610, D5S1416, D5S107, D5S1356 and D5S57. The molecular polymerase chain reaction technique is used for analyzing allele segregation, which allows to identify affected chromosomes by obtaining genotypes in families diagnosed with types 1, 2 and 3 SMA. It was determined that in a family whose eldest son had type 3 SMA, his 4 years-old sister was a SMA gene carrier whereas his 5 years-old brother was helathy. genetic counselling was given to this family.
Subject headings: ENFERMEDAD DE WERDNING-HOFFMAN/genetics;WERDNING HOFFMANN DISEASE/diagnosis; CUBA.
1 Laboratorio de Genética. Instituto de Neurología y Neurocirugía, Ciudad de La Habana.
2 Unidad de Genética Molecular. Hospital Ramón y Cajal, Madrid, España. ]]>