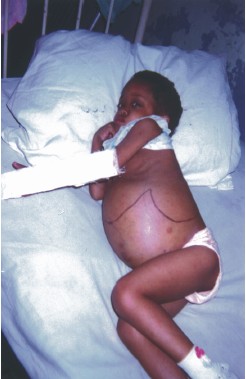
Fig.1. Gran hepatoesplenomegalia y ascitis en una niña de 4 años de edad con histiocitosis de células de Langerhans multisistémica
Instituto de Hematología e Inmunología
Dra. Eva Svarch, Dr. Rafael Arteaga, Dra. Valia Pavón Morán y Dr. Alejandro González Otero
El término histiocitosis identifica un grupo de alteraciones que tienen en común la proliferación de células dendríticas (CD) y los macrófagos, y se diagnostican más frecuentemente en niños. Dentro de las relacionadas con las CD las fundamentales son las histiocitosis a células de Langerhans (HCL). Las HCL tienen un comportamiento clínico muy variable, que puede ir desde una lesión que involucra un solo sitio o sistema hasta una enfermedad multisistémica. El tratamiento depende de la extensión del proceso. Una lesión única tiende a desaparecer espontáneamente. También la biopsia diagnóstica con o sin inyección de un esteroide puede iniciar la curación. Los pacientes con enfermedad multisistémica pueden beneficiarse con el tratamiento esteroideo y citostático o inclusive con el trasplante de células progenitoras hematopoyéticas. La histiocitosis sinusal con linfoadenopatías masivas o enfermedad de Rosai Dorfman se debe a la proliferación de los macrófagos, es de naturaleza benigna y usualmente autolimitada. Afecta sobre todo a niños y adultos jóvenes. La linfohistiocitosis hemofagocítica también se produce por la proliferación de los macrófagos y es una enfermedad rara con una alta mortalidad. Puede ser familiar (autosómica recesiva) o secundaria a infecciones virales. Esta última forma se presenta más frecuentemente en el lactante pequeño. En la actualidad, sobre todo en la variedad familiar, el trasplante alogénico de células progenitoras hematopoyéticas puede ser la única medida curativa.
]]>
DeCS: HISTIOCITOSIS DE CELULAS NO LANGERHANS/diagnóstico; HISTIOCITOSIS DE CELULAS DE LANGERHANS/diagnóstico; HISTIOCITOSIS DEL SENO/diagnóstico; NIÑO.
Las histiocitosis son enfermedades del sistema histiofagocítico que se presentan más frecuentemente en los niños.
El histiocito es una célula del sistema inmune que incluye, entre otros, a los macrófagos y a las células dendríticas o dendrocitos. Los macrófagos son los encargados de procesar el antígeno y las células dendríticas de presentarlo a los linfocitos T. Una de las células dendríticas más importante es la célula de Langerhans (CL) que tiene su origen en la médula ósea y reside, en condiciones normales, en la piel, mucosas malpighianas y pulmón; representa del 1 al 2 % de las células de la epidermis y es importante en la vigilancia inmunológica cutánea.
La clasificación actual de las histiocitosis se expone en el anexo 1.1
En este trabajo nos referiremos solamente a las histiocitosis relacionadas con las CL, y a algunas relacionadas con los macrófagos, la enfermedad de Rosai Dorfman y la histiocitosis hemofagocítica.
Conocida anteriormente como histiocitosis X (granuloma eosinófilo, enfermedad de Letterer Siwe y enfermedad de Hans Schuller Christian),2 tiene un espectro clínico muy amplio, que va desde una lesión osteolítica que cura espontáneamente hasta una enfermedad letal semejante a la leucemia. Su evolución es extremadamente variable; indolente durante mucho tiempo o rápidamente progresiva y fatal. También una lesión única puede evolucionar hacia una forma diseminada o hacia la cronicidad. Puede presentarse a cualquier edad, desde el nacimiento hasta la ancianidad, pero se diagnostica más entre 1 y 13 años y es ligeramente más frecuente en varones.3
]]> Su incidencia es de 0,54 / 100 000 niños de 0 a 15 años y de 1,64 / 100 000 en niños entre 0 y 2 años de edad.4
Etiopatogenia
A pesar de numerosos estudios, la etiología es aún desconocida. La etiología viral es poco probable, aunque en algunos pacientes se ha encontrado el genoma de un herpes virus tipo 6. Tampoco se ha podido comprobar que sea una alteración inmunológica primaria. Se ha planteado la estimulación de los linfocitos T por un superantígeno, pero esto no se ha podido corroborar.5
Las CL tienen su origen en las células CD34 positivas de la médula ósea, de la cual migran por vía hematógena hacia la epidermis cubriendo con sus dendritas el 25 % de su superficie.6 A partir de la piel migran por vía linfática hacia la zona paracortical de los ganglios para cumplir su función de presentación del antígeno a los linfocitos T. Al contrario de los monocitos y macrófagos, su función de presentación del antígeno predomina sobre la actividad fagocítica.
En la histiocitosis a células de Langerhans (HCL) las CL pueden infiltrar todos los órganos: hígado, bazo, tracto digestivo, pulmón, sistema nervioso central y hueso. Es probable que en su migración participen moléculas de adhesión específicas. Se ha encontrado expresión de los CD54, CD58, b1 integrina, a 4, CD2, CD11a, CD11b y CD62L.7
Algunas manifestaciones generales como fiebre y pérdida de peso parecen estar condicionadas por citocinas: IL 1, IL8; FNT a, GM-CSF, interferón g.7,8
A pesar de que se ha demostrado que se trata de un proceso clonal, la mayoría de los autores coinciden en que es de naturaleza reactiva y no neoplásica.9 ]]>
Anatomía patológica
En contraste con la heterogeneidad clínica de la HCL, los hallazgos histológicos son uniformes en todas las variedades; el más característico es la presencia de la CL en un contexto semejante al de la inflamación con neutrófilos, eosinófilos, células plasmáticas, linfocitos e histiocitos multinucleados gigantes.10
El hallazgo morfológico patognomónico de las CL es la presencia, en el estudio con microscopia electrónica, de los corpúsculos de Birbeck en forma de raqueta localizados en el citoplasma, adyacentes o contiguos a la membrana.11 Inicialmente las lesiones son celulares y en ellas predominan los histiocitos, pero cuando tienen mucho tiempo de evolución, sobre todo las óseas, son paucicelulares, fibróticas, algunas semejantes al xantogranuloma.10
Caracterización de la CL
]]>
Las CL tienen marcadores de membrana que reaccionan con la aglutinina del maní, la fosfatasa alcalina placentaria, el receptor para el interferón a, la ATP asa, la a D manosidasa,12 el CD45 y la proteína S-100.10,13 Un marcador muy específico es el CD1a, que puede ser positivo también en algunos casos de xantogranuloma y de enfermedad de Rosai Dorfman.2 En la actualidad este marcador se puede estudiar en láminas procesadas con parafina mediante el CD 010.14,15
Diagnóstico
El diagnóstico puede ser presuntivo cuando el cuadro clínico y la morfología con microscopia óptica son característicos; probable cuando a las lesiones histológicas típicas se le agrega la positividad por lo menos de 2 de los siguientes marcadores: ATPasa, lectina del maní, proteína S-100 y a D manosidasa, y definitivo cuando hay positividad al CD1a y están presentes los gránulos de Birbeck en la microscopia electrónica.1
Hay autores que consideran que cuando el cuadro clínico y radiológico es típico, se puede realizar el diagnóstico con un estudio histológico por microscopia óptica con la coloración de hematoxilina eosina.5
Cuadro clínico
Depende de la extensión de la enfermedad y del tejido u órgano comprometido. En la enfermedad multisistémica están afectados muchos órganos y pueden presentarse manifestaciones generales: fiebre, anorexia, pérdida de peso, anemia, manifestaciones hemorrágicas (sobre todo petequias localizadas en tronco fundamentalmente), astenia e irritabilidad.16 ]]>
Lesiones óseas
Se encuentran en casi todos los pacientes con enfermedad localizada y son muy frecuentes en la multisistémica.
El signo inicial suele ser un aumento de tamaño indoloro de las partes blandas. El cráneo es el sitio más afectado, en segundo lugar los huesos largos de los miembros superiores y luego los planos: costillas, pelvis y vértebras.
El estudio radiológico muestra una o varias lesiones líticas de bordes bien delimitados. Puede existir exoftalmía cuando se afectan las paredes de la órbita.
Las lesiones que se producen en la mastoides son semejantes a la mastoiditis y si el proceso se extiende al oído medio puede conducir a la sordera. Cuando la osteólisis tiene lugar en el maxilar se producen dientes flotantes. En una niña con HCL multisistémica atendida en nuestro instituto se observó una necrosis digital no descrita con anterioridad producida por lesiones de vasculitis.17
]]>
Piel
Las manifestaciones cutáneas son muy frecuentes y a menudo el primer signo de la enfermedad; son lesiones eritematosas y escamosas parecidas a la dermatitis seborreica. Se localizan en la piel del cráneo, cara, regiones retroauriculares, pliegues y región perianal. En el lactante sin localizaciones de la HCL en otros sitios, pueden curar espontáneamente.18
Médula ósea y sangre periférica
La anemia y la trombocitopenia no son raras, menos frecuente es la leucopenia. Estas alteraciones se atribuyen a una disfunción de la médula ósea, pero su patogenia no es clara. En la médula ósea normal no parecen existir CL, aunque sí otros tipos de células dendríticas. El aumento de histiocitos no es diagnóstico y no es frecuente que se encuentre infiltración, por lo que su estudio no es concluyente.10 Cuando se demuestra infiltración se acompaña casi siempre de gran hepatoesplenomegalia, lesiones de la piel y fiebre, y tiene valor pronóstico desfavorable.19 Recientemente se ha comenzado el estudio del CD1a en la médula ósea por citometría de flujo.20
]]>
Hígado y bazo
El hígado puede estar aumentado de tamaño por infiltración por CL o debido a la compresión de la vena porta por ganglios linfáticos. La hepatomegalia puede también relacionarse con la hiperplasia e hipertrofia de las células de Kupffer, como resultado de la activación generalizada del sistema inmunológico celular sin infiltración por CL ni hepatopatía obstructiva. Las alteraciones histopatológicas oscilan desde la colestasis moderada hasta la severa infiltración por CL, CD1a +, pero sin gránulos de Birbeck,18 de las áreas portales con evidencias de lesiones hepatocelulares y de los conductillos biliares que pueden progresar hacia la colangitis esclerosante, cirrosis biliar e insuficiencia hepática. Ésta se manifiesta por ictericia, edemas por hipoalbuminemia y alteraciones de la coagulación.
El aumento de tamaño del bazo puede ser un factor adicional en las citopenias producidas por el hiperesplenismo secundario.
En una paciente de nuestro servicio que no respondió al tratamiento es evidente la gran hepatoesplenomegalia con ascitis (fig. 1).
Fig.1. Gran hepatoesplenomegalia y ascitis en una niña de 4 años de edad con histiocitosis de células de Langerhans multisistémica
Pulmones
Las lesiones pulmonares ocurren a cualquier edad, pero son más frecuentes en la tercera década de la vida. Generalmente se acompañan de fiebre, disnea y pérdida de peso. En la radiografía de tórax aparece un infiltrado micronodular y el diagnóstico se realiza por la presencia de células CD1a positivas en el líquido del lavado bronquial. ]]>
Evolutivamente aparecen en ocasiones quistes o bulas que al romperse producen neumotórax. En la fase final existen fibrosis y enfisema.21
Aparato gastrointestinal
Las lesiones del aparato gastrointestinal son raras. Se evidencian por un síndrome de malabsorción con detención del crecimiento, diarreas con o sin sangre o enteropatía exudativa. En el estudio radiológico se observan segmentos de dilatación del intestino delgado que alternan con otros de estenosis. Para el diagnóstico es necesario realizar endoscopia y biopsia de la mucosa intestinal.18,22
Timo
]]>
Puede haber aumento de tamaño del órgano y en ocasiones es el único afectado. En la necropsia se encuentra siempre infiltrado.23
Glándulas endocrinas
La alteración más frecuente es la diabetes insípida, y aunque en general aparece en períodos avanzados de la evolución o como una secuela, se puede presentar en cualquier período en el 17,5 % de los pacientes.3
Su patogenia no se conoce bien y el diagnóstico presuntivo se realiza por la prueba de deprivación de agua durante 7 horas y se confirma por la medición de la vasopresina en orina.24
La diabetes insípida aparece fundamentalmente cuando existen lesiones en el cráneo o enfermedad multisistémica.
]]>
Sistema nervioso central
Pocos pacientes presentan alteraciones del sistema nervioso central, su frecuencia exacta se desconoce. Además de las lesiones del hipotálamo con la consecuente diabetes insípida, se describen alteraciones del cerebelo, tallo cerebral, hemisferios cerebrales y médula espinal.
Algunos enfermos presentan signos de hipertensión intracraneal o ataxia, adiadococinecia, temblor, disartria, hiperreflexia, hemiplejía o cuadriplejía y disfagia, con o sin déficit intelectual.25
Asociación de la HCL con enfermedades malignas
Su frecuencia es mayor que lo que cabría esperar por el azar.26 La asociación con leucemias, linfomas o tumores sólidos puede ser atribuida a la quimioterapia o a la radioterapia utilizada en la HCL, pero en ocasiones la precede y a veces coexisten ambos procesos.26
]]>
Pronóstico
Depende de la edad en el momento del diagnóstico y del cuadro clínico inicial. Los niños con menos de 2 años de edad y disfunción hepática, pulmonar o de la médula ósea, tienen mal pronóstico. Los aspectos que se tienen en cuenta para el criterio de mal pronóstico en la HCL se detallan en el anexo 2.27 Existe una correlación lineal entre la mortalidad y el número de órganos afectados; cuando 1 ó 2 están tomados, la mortalidad es del 10 %, cuando son más de 2 la mortalidad alcanza el 90 %.28 Los pacientes con enfermedad localizada en hueso tienen un excelente pronóstico, con mortalidad casi nula. Parece ser que en un niño con enfermedad multisistémica la presencia de lesiones óseas tiene buen pronóstico, mientras que las lesiones cutáneas son de pronóstico desfavorable.29
Tratamiento
Depende de la extensión de la lesión. Cuando está localizada a un solo tejido, habitualmente hueso, se puede observar su evolución sin realizar ningún tratamiento. El simple curetaje con o sin inyección de esteroides, cura del 80 al 90 % de los casos; en el 10 % de ellos se producen recidivas, nuevas localizaciones o secuelas, como la diabetes insípida.30
La radioterapia solo se debe utilizar cuando la lesión es inaccesible a la cirugía y compromete órganos críticos, por ejemplo vértebras, con posible compresión de la médula espinal. Las dosis deben ser bajas: de 400 a 800 cGy. ]]>
Cuando la enfermedad afecta un solo sistema en muchos sitios o es multisistémica, se debe emplear la quimioterapia.El primer estudio cooperativo que demostró la eficacia de la quimioterapia aplicada temprano en la evolución de la enfermedad y que permitió el análisis de niños con diferentes grados de actividad, fue el DAL HX-83.31 En 1991, la Sociedad Internacional del Histiocito instituyó el protocolo LCH-I, en el cual los pacientes se dividieron al azar para recibir prednisona y vinblastina (VBL) o prednisona y etopósido (VP 16). Con este tratamiento se pudieron identificar en las primeras 6 semanas a los enfermos que no respondían favorablemente, los que se pasaron a la otra rama. Sin embargo, del 10 al 20 % de los enfermos desarrollaron enfermedad progresiva y fallecieron.32
El tratamiento que recomienda en la actualidad la Sociedad Internacional del Histiocito es el LCH-II que se expone en las figuras 2 y 3.20 En este esquema se recomienda utilizar la VBL a 0,2 mg/kg de peso en los niños de menos de 10 kg y profilaxis de la infección por Neumocistis carinii con sulfametoxazol-trimetropín. Los pacientes de bajo riesgo o con enfermedad localizada que requieren quimioterapia se tratan todos con la rama A.20 En los enfermos que no responden a ninguna de las ramas de ese protocolo se aplica el protocolo LCH-S, que consiste en la realización de trasplante de células progenitoras hematopoyéticas20 o tratamiento inmunosupresor con ciclosporina A y globulina antitimocítica.33
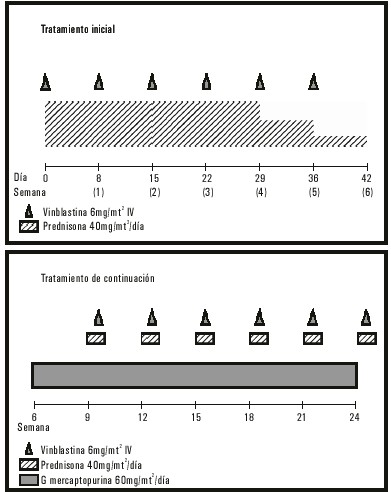
Fig. 2. Protocolo HCL-II Rama A.
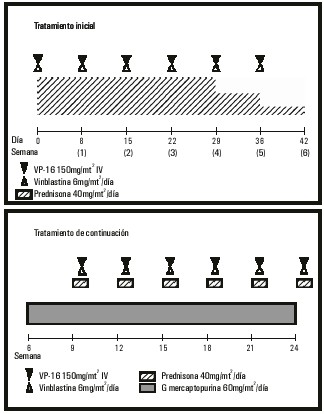
Fig. 3. Protocolo HCL-II Rama B.
A diferencia de lo que ocurre en el cáncer, el objetivo del tratamiento en la HCL es el control y no la erradicación de todas las lesiones; el término remisión completa, por lo tanto, no se debe usar.34
Existen varios tipos de respuesta: la más favorable es aquella en la que no se demuestran evidencias de enfermedad con desaparición de los signos y síntomas. En otras ocasiones los signos y síntomas persisten o mejoran, sin aparición de nuevas lesiones, y en otras la HCL progresa, ya sea porque empeoran los signos y síntomas detectados en el momento del diagnóstico o porque aparecen otros nuevos.
Algunos aspectos del tratamiento de la HCL aún no están claros. Hay acuerdo en cuanto a cómo tratar una lesión ósea única y en cuanto a que en una enfermedad con compromiso multisistémico una quimioterapia relativamente agresiva es beneficiosa, pero no hay consenso en cómo tratar la enfermedad progresiva refractaria, la que incluye la hipófisis con diabetes insípida, la crónica que recae y la infiltración crónica y progresiva del pulmón, hígado o sistema nervioso central. Parte de esa falta de consenso se debe a que persiste la ambivalencia en relación con la patogenia, pues aún no está definido si es una enfermedad neoplásica, una alteración inmunológica o ambas.35
Es más frecuente en adultos jóvenes. Su etiología es desconocida. Se ha relacionado con una infección por el virus de Epstein-Barr, lo que podría alterar la respuesta a un antígeno específico pero desconocido.36 Existen evidencias de que la célula infiltrante es de naturaleza policlonal.37
Se presenta en general con adenopatías cervicales bilaterales, a veces desfigurantes indoloras (fig. 4). Cualquier grupo ganglionar puede estar afectado. En el 30 % de los enfermos las manifestaciones pueden ser extraganglionares, las más frecuentes son: nasofaringe, glándulas salivales, cavidad oral, huesos y piel.36
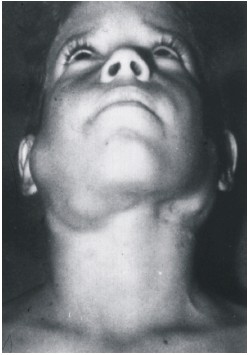
Fig. 4. Adenopatías cervicales en un paciente de 14 años con enfermedad de Rosai-Dorfman.
La inmunopatología de esta entidad consiste en un llenado progresivo de los sinusoides de los ganglios linfáticos con histiocitos y linfocitos que llevan casi a un borramiento de la arquitectura del ganglio linfático.
Elementos muy importantes son la leucofagocitosis y la eritrofagocitosis.
En ocasiones el paciente presenta fiebre y corta estatura.36
Las lesiones cutáneas son xantomatosas y las del hueso, líticas, difíciles de diferenciar de las de la HCL.
No se ha definido aún un tratamiento estándar. Debido a que casi siempre tiene un curso autolimitado se trata de no usar quimioterapia. Por otra parte, ni ésta ni los esteroides han probado su eficacia. Algunos pacientes han respondido al interferón a y otros a la G mercaptopurina y methotrexato (MTX) orales administrados durante largos períodos de tiempo. ]]>
Si la enfermedad es indolente no se aconseja tratamiento.
La linfohistiocitosis hemofagocítica (LHHF) puede ser familiar, autosómica recesiva, o secundaria. La LHHF familiar se sospecha cuando algún otro miembro de la familia ha sido afectado, o cuando se comprueba consanguinidad en los padres, además se presenta en los primeros meses de la vida siempre en niños menores de 2 años y es uniformemente fatal. La variedad secundaria aparece después de los 2 años, parece estar relacionada con infecciones virales, sobre todo con el virus de Epstein-Barr (síndrome hemofagocítico asociado a virus), y puede curar.38
La frecuencia de las manifestaciones clínicas en la LHHF se observa en la figura 5 y en la 6 se expone el porcentaje de los resultados de las pruebas de laboratorio.
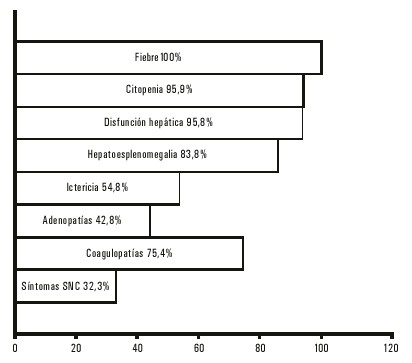
Fig. 5. Linfohistiocitosis hemofagocítica. Manifestaciones clínicas.
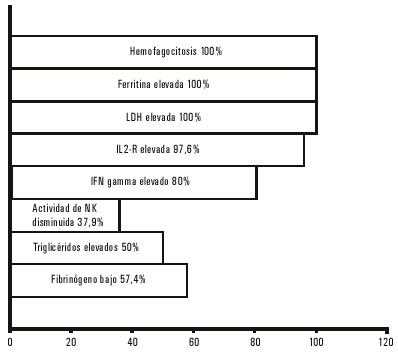 ]]>
]]>
En los 2 tipos de LHHF la patogenia se desconoce. Se plantea que está en relación con un defecto en la activación de las células asesinas naturales. Las interacciones inmunológicas entre las células son exageradas y persistentes. En algunos casos el proceso es monoclonal.39
Los hallazgos patológicos típicos son: proliferación marcada de histiocitos sin características de malignidad con marcada hemofagocitosis.12 La infiltración por histiocitos y linfocitos es generalizada, abarca la médula ósea, los órganos linfoides secundarios y todos los órganos vitales, incluyendo el sistema nervioso central.40,41
Sin tratamiento, la LHHF primaria es rápidamente fatal. Se han utilizado diferentes protocolos en los que los medicamentos de elección son la predinosona con VBL o con VP16. La radioterapia en cráneo y el MTX intratecal han aumentado la sobrevida. Exanguino-transfusiones o plasmaféresis repetidas han inducido remisión en algunos pacientes, al igual que el uso de globulina antitimocítica y de la ciclosporina A. Estos tratamientos han logrado aumentar la sobrevida, a veces hasta los 5 años, pero no la curación. El único tratamiento curativo es el trasplante alogénico de células progenitoras hematopoyéticas. La Sociedad Internacional del Histiocito recomienda el protocolo HLA-94, en el que se combina la ciclosporina A con prednisona y VP16 y MTX intratecal. El objetivo es conseguir la estabilización del cuadro clínico para realizar después el trasplante de médula ósea.
En la LHHF asociada a virus se recomendaba evitar la quimioterapia. Sin embargo, hay casos controlados con quimioterapia y también se han obtenido éxitos con el trasplante de células proge-nitoras hematopoyéticas, sobre todo cuando se demuestra que la activación de las células asesinas naturales está disminuida o ausente. Una extensa revisión sobre este tema se ha publicado recientemente.42
Anexo 1. Clasificación actual de las alteraciones histiocíticas
Alteraciones con comportamiento biológico variable
]]>
- Relacionadas con la célula dendrítica:• Histiocitosis de células de Langerhans.
• Procesos a células dendríticas secundarios.
• Xantogranuloma juvenil y alteraciones asociadas.
• Histiocitomas solitarios de células dendríticas con fenotipos variables.
- Relacionadas con el macrófago:
• Síndromes hemofagocíticos. ]]>
• Linfohistiocitosis hemofagocítica primaria (familiar y esporádica producida por infecciones virales).
• Síndromes hemofagocíticos secundarios.
Asociados a infección.
Asociados a procesos malignos.
Otros.
• Enfermedad de Rosai Dorfman (histiocitosis sinusal con adenopatías masivas).
• Histiocitoma solitario con fenotipo de macrófago.
Alteraciones malignas
- Relacionadas con el monocito: ]]>
• Leucemias (clasificación FAB).
• Leucemia monocítica M5a y b.
• Leucemia mielomonocítica M4.
• Leucemia mielomonocítica crónica.
• Tumor o sarcoma monocítico extramedular (contraparte monocítica del sarcoma granulocítico).
- Relacionadas con células dendríticas:
]]>• Sarcoma histiocítico relacionado con la célula dendrítica (localizado o diseminado) de fenotipo específico: célula dendrítica folicular, célula dendrítica interdigitante, etc.
- Relacionadas con el macrófago:
• Sarcoma histiocítico relacionado con el macrófago (localizado o diseminado).
Anexo 2. Criterios de mal pronóstico de la HCL
Alteraciones hepáticas ]]>
-Proteínas totales < 5,5 g/dL
Alteraciones pulmonares
-Taquipnea
-Disnea
-Cianosis ]]>
Alteraciones hematopoyéticas
-Hemoglobina < 10 g/dL
-Leucocitos < 4 x 109 /L
-Neutrófilos < 1,5 x 109 /L
-Plaquetas < 100 x 109 /L
]]>
The term histiocytosis identifies a group of disorders that have in common the proliferation of dentritic cells (DC) and macrophages and is frequently diagnosed in children. Among the fundamental variants of histiocytosis related with DC, we find Langerhans cell histiocytosis (LCH). Langerhans cell histiocytosis has very variable clinical behavior that ranges from a lesion involving only one site or system to a multisystem disease. Treatment depends on the spread of the process. An only lesion tends to spontaneously disappear. Also diagnostic biopsy with or without steroid injection may lead to healing. Those patients with multisystem disease may benefit from an steroid and cytostatic-based treatment or even from progenitor hematopoietic cell transplantation. Sinus histiocytosis with massive lymphadenopathies or Rosai Dorfman disease is a benign and usually self-limited disease which is caused by the macrophage proliferation; it generally affects children and young adults. Hemophagocytic lymphohistiocytosis is also caused by macrophage proliferation and is a rare disease with a high mortality rate. It can be familiar hemophagocytic lymphohistiocytosis (recessive autosomal) or secondary to viral infections, being the latter form the most frequent in infants. At present, mainly in the familiar variant, the progenitor hematopoietic allogenic transplant may serve as the only curative option.
Subject headings: HISTIOCYTOSIS, NON-LANGERHANS-CELL/diagnosis; HISTIOCYTOSIS LANGERHANS-CELL /diagnosis; HISTIOCYTOSIS, SINUS/diagnosis; CHILD.
1. Favara BE, Feller AC, Pauli M, Jaffe ES, Weiss LM, Aricó M, et al. A contemporary clasification of histiocytic disorders of childhood. Med Pediatr Oncol 1997;29:157-66.
2. Favara BE. Langerhans cell histiocytosis: pathobiology and pathogenesis. Semin Oncol 1991;18:3-7.
3. The French Langerhans cell histiocytosis study group: 348 cases observed between 1983 and 1993. Arch Dis Child 1996;75:17-24.
4. Catersen H, Ornvold K. The epidemiology of Langerhans cell histiocytosis in children in Denmark 1975-1984. Med Pediatr Oncol 1993;21:387-8.
5. Beverley PCL, Abbas AK. The scientific challenge of Langerhans cell histiocytosis. Br J Cancer 1994;70:561-3.
6. Chu T, Jaffe R. The normal Langerhans cell and the LCH cell. Br J Cancer 1994;70:4-10.
7. Graaf J, Rienk Y, Tamminga J, Kamps WA, Timens W. Expression of cellular adhesion molecules in Langerhans cell histiocytosis and normal Langerhans cell. Am J Pathol 1995;147:1161-71.
8. Kannouraris G, Abbas A. The role of citokines in the pathogenesis of Langerhans cell histiocytosis. Br J Cancer 1994;70:s37-40.
9. Willman C, Busque L, Griffith B. Langerhans cell histiocytosis (histiocytosis X). A clonal proliferation disease. N Eng J Med 1994;331:154-60.
10. Schmitz L, Favara BE. Nosology and pathology of Langerhans cell histiocytosis. Hematol Oncol Clin North Am 1998;12:221-46.
11. Favara BE, Jaffe R. Pathology of Langerhans cell histiocytes. Hematol Oncol Clin North Am 1987;1:75-97.
12. Histiocyte Society Writing Group. Histiocytosis syndrome in children. Lancet 1987;1:208-9.
13. Thomas C, Donnadieu J, Emile JF, Brouse N. Groupe dÉtude sur l´Histiocytosis lanhergansienne de la Societé d´Hematologie et d´Inmunologie Pediatriques. Arch Pediatr 1996;3:63-9.
14. Krenacs L, Yidzaviez Y, Boumsell J. Immunohistochemical detection of Cd1 a antigen in formalin fixed and paraffin embedded tissue sections with monoclonal antibody 010. J Pathol 1993;171:99-104.
15. Kilpatrick SE, Wenger DE, Gilchrist GS, Shives TC, Wolland PC, Unni KK. Langerhans cell histiocytosis (Histiocytosis X)of bone. A clinicopathogic analysis of 263 pediatric and adults cases. Cancer 1995;76:2471-84.
16. Clinical Writing Group of the Histiocyte Society. Histiocytosis syndrome in children II: approach to the clinical and laboratory evaluation of children with Langerhans cell histiocytosis. Med Pediatr Oncol 1989;17:492-5.
17. Svarch E, Crombet O, González A, Machín S, García T, Cabrera H, et al. Digital necrosis associated with Langerhans ell histiocytosis. Med Pediatr Oncol 1988;28:31-32.
18. Egeler RM, D´Angio GJ. Langerhans cell histiocytosis. J Pediatr 1995;127:1-11.
19. Egeler RM, Nesbit ME. Langerhans cell histiocytosis and others disorders of monocyte-histiocyte lineage. Crit Rev Oncol Hematol 1995;18:9-35.
20. Gadner H, Ladish S, Broadbent V. Histiocyte Society. LCH-II Langerhans cell histiocytosis. Treatment protocol of the Second International Study and LCH-S Langerhans cell histiocytosis. Treatment protocol of the international study for severe progressive LCH (Salvage) International Study, Vienna, 1996.
21. Travis WD, Borok Z, Roum JH, Zhang J, Feurerstein I, Ferans VJ, et al. Pulmonary Langerhans cell granulomatosis (Histiocytosis X); a clinicopathologic study of 48 cases. Am J Pathol 1993;17:971-86.
22. Egeler RM, Schipper MEI, Heymans HSA. Gastrointestinal involvement in Langerhans cell histiocytosis (histiocytosis X): a clinical report of three cases. Eur J Pediatr 1990;149:325-9.
23. Hamoudi AB, Newton WA, Mancer K, Penn GM. Thymic changes in histiocytosis. Am J Clin Pathol 1982;77:169-74.
24. Broadbent V, Pritchard J. Diabetes insipidus associated with Langerhans cell histiocytosis: Is it reversible? Med Pediatr Oncol 1997;28:289-93.
25. Grois N, Barkovich J, Rosenau W, Ablin AR. Central nervous system disease associated with Langerhans cell histiocytosis. Am J Pediatr Hematol Oncol 1993;15:245-54.
26. Egeler RM, Neglia JP, Arico M, Favara BE, Heitger A, Nesbit ME, et al. A acute leukemia in association with Langerhans cell histiocytosis. Med Pediatr Oncol 1994;23:81-5.
]]>27. Lahey ME. Prognosis in reticuloendotheliosis in children. J Pediatr 1962;60:664-6.
28. Histiocytosis X-An analysis of prognostic factors. J Pediatr 1975;87:184-8.
29. Broadbent V. Favourable prognosis in histiocytosis X: bone involvement and absence of skin disease. Arch Dis Child 1986;61:1219-21.
30. Greenberger JS, Crocker AC, Vawter G, Jaffe N, Cassady JR. Results of treatment of 127 patients with systemic histiocytosis (Letterer Siwe syndrome Schuller Christian syndrome and multifocal eosinophilic granuloma). Medicine 1981;60:311-38.
31. Gadner H, Heitger A, Grois N, Gatterer-Menz I, Ladisch S. A treatment strategy for disseminated Langerhans cell histiocytosis. Med Pediatr Oncol 1994;23:72-80.
32. Ladisch S, Gadner H. Treatment of Langerhans cell histiocytosis Evolution and current approaches. Br J Cancer 1994;70(Suppl 23):541-6.
33. Bertrand Y. Traitement des formes sevéres d´histiocytose langerhansienne. Pediatrie 1993;48:770-4.
34. Komp DM. Concepts in staging and clinical studies for treatment of Langerhans cell histiocytosis. Semin Oncol 1991;18:18-23.
35. Arceci RJ, Brenner MK, Pritchard J. Controversies and new approaches to treatment of Langerhans cell histiocytosis. Hematol Oncol Clin North Am 1998;12:339-57.
36. Foucar E, Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy (Rosai Dorfman disease); review of the entity. Semin Diag Pathol 1990;7:19-73.
37. Paulli M, Bergamaschi G, Tonon L, Viglio A, Rosso R, Facchetti F, et al. Evidence for a polyclonal nature of the cell infiltratet in sinus histiocytosis with massive lymphadenopathy (Rosai Dorfman disease). Br J Haematol 1995;91:415-8.
38. Risdall RJ, Mc Kenna RW, Nesbit ME, Krivit W, Balfour HH, Simmons RL, et al. Virus associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from malignant histiocytosis. Cancer 1979;44:993-1002.
39. Imashuku S, Hibi S, Todo S. Hemophagocytic limphohistiocytosis in infancy and childhood. J Pediatr 1997;130:352-7.
40. Filipovich AH. Hemophagocytic lymphohistiocytosis: a lethal disorder of inmune regulation. J Pediatr 1997;130:337-8.
41. Henter JI, Elinder G. Cerebromeningeal haemophagocytic lymphohistiocytosis. Lancet 1992;339:104-7.
42. Henter JI, Aricó M, Elinder G, Imashuku S, Janka G. Familiar hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am 1998;12:417-33.
Recibido: 12 de octubre del 2000. Aprobado: 24 de noviembre del 2000.
Dra. Eva Svarch. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800, Ciudad de La Habana, Cuba. Teléf: (537)578268. Fax: (537)338979. e-mail:ihidir@hemato.sld.cu ]]>