
Instituto de Hematología e Inmunología
Lic. Mayelín Herrera García y Dra. Marianela Estrada del Cueto
La esferocitosis hereditaria (EH) es una enfermedad caracterizada por anemia hemolítica de severidad variable, con presencia de esferocitos en sangre periférica y una respuesta clínica favorable a la esplenectomía. Con el desarrollo de nuevas técnicas se encontraron las primeras alteraciones bioquímicas de las proteínas de la membrana eritrocitaria, y posteriormente, se han podido precisar las alteraciones moleculares mediante las técnicas del ADN recombinante. La EH es una enfermedad muy heterogénea que se produce por un defecto intrínseco del glóbulo rojo, y existen otras alteraciones secundarias a esta afección. La prueba más utilizada para el diagnóstico de la EH es la fragilidad osmótica del glóbulo rojo. Se ha demostrado que esta enfermedad es producida por defectos de las proteínas que intervienen en las interacciones verticales entre el esqueleto de la membrana y la bicapa lipídica. El tratamiento de elección en la EH es la esplenectomía, ya que es el más efectivo en el control de la anemia, aunque la sobrevida de los glóbulos rojos permanece acortada y los esferocitos no desaparecen. Este proceder se indica en pacientes con anemia hemolítica severa o en individuos moderadamente asintomáticos pero que presentan litiasis vesicular.
DeCS: ESFEROCITOSIS HEREDITARIA/diagnóstico; ESFEROCITOSIS HEREDITARIA/ genética; ADN RECOMBINANTE; PROTEINAS DE LA MEMBRANA/química; PROTEINAS RECOMBINANTES/química; ESPLENECTOMIA; MEMBRANA ERITROCITICA/química.
La membrana del glóbulo rojo es la responsable de las propiedades mecánicas y de la mayoría de las funciones fisiológicas de la célula.1
Está formada por una bicapa lipídica plana, donde predominan en el 80 % los fosfolípidos y el colesterol y en menor medida los glicolípidos y aminofosfolípidos, distribuidos asimétricamente. De igual forma, se encuentran embebidas parcial o totalmente en ella las proteínas integrales de membrana, unidas fuertemente por enlaces apolares. Su libre desplazamiento a través de esta bicapa contribuye a mantener su fluidez.2 Las proteínas periféricas interactúan entre sí para formar una malla o enrejado que recubre la cara interior de la doble capa de fosfolípidos y son las responsables de la estabilidad y las propiedades viscoelásticas de la membrana.3 Entre estas proteínas se destacan la espectrina (Sp), la ankirina (banda 2.1, 2.2, 2.3 y 2.6), la banda 4.1, la banda 4.2, la banda 4.9, la aducina, la tropomiosina y la banda 7. Otras proteínas periféricas se disponen hacia la cara exterior de la bicapa lipídica y ellas son fundamentalmente antígenos de grupo sanguíneo4 (fig.1).
Fig. 1. Estructura de la membrana eritrocitaria.
La banda 3 representa el 25 % del total de las proteínas integrales. Está constituida por 2 dominios estructurales: el dominio citoplasmático, que es el encargado de la unión con las proteínas del esqueleto, y el sitio transmembranoso que mantiene el contacto con el medio extra e intracelular, proporcionando los canales responsables del transporte de iones bicarbonato (HCO3-) y cloruros (CI-). Además, posee un sitio de glicosilación capaz de unir antígenos para el grupo sanguíneo I/i5 e interviene activamente en la eliminación de eritrocitos envejecidos.6
]]> Las glicoforinas son un grupo de proteínas integrales caracterizadas por su elevado contenido en ácido siálico. Las glicoforinas A,B,C y D son las más importantes y constituyen los sustratos antigénicos de los diferentes grupos sanguíneos. La glicoforina C contribuye a la estabilidad de la membrana gracias a su interacción con proteínas periféricas, además de participar en el intercambio iónico transmembranoso.7La Sp es la proteína más abundante y además la principal responsable del mantenimiento del enrejado proteico.8 Está compuesta por 2 subunidades a y b enrolladas de forma antiparalela, las que se unen por sus extremos para formar tetrámeros. Estas 2 subunidades están constituidas por secuencias repetitivas de 106 aminoácidos, las que se enlazan para formar una triple hélice9 (fig.2).
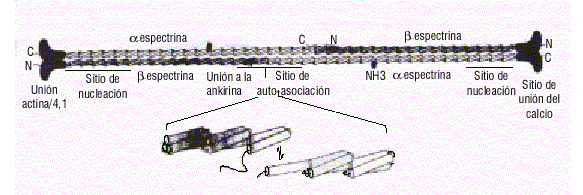
Fig.2. Estructura del tetrámero de espectrina (Sp). Se observa la triple hélice de las unidades repetitivas, los sitios de autoasociación de la Sp y los sitios de nucleación donde comienza la interacción lateral entre las cadenas a y b de la Sp.
La ankirina (ANK) está compuesta por 3 subunidades estructurales correspondientes a 3 dominios funcionalmente diferentes: regulador, de unión a la membrana y de unión a la Sp. Su contribución a la integridad de la membrana es decisiva, ya que constituye un importante punto de anclaje a la doble capa lipídica a través de la banda 3.10
La actina es una proteína organizada en forma de protofilamentos helicoidales estabilizados por la interacción con la Sp, la proteína 4.1 y la tropomiosina.11
Otra de las proteínas que integran el citoesqueleto es la proteína 4.1, cuya función fundamental es estabilizar la unión espectrina-actina y contribuir a fijar el esqueleto a la bicapa lipídica.12
El mantenimiento de la forma y estabilidad de la membrana es también responsabilidad de otras proteínas. Entre ellas está la proteína 4.2, que actúa como modulador para estabilizar la interacción ankirina-banda 3.13 La banda 4.9, la aducina y la tropomiosina, protegen la estabilidad de la actina.14
Los estudios moleculares han permitido conocer la localización cromosómica de estas proteínas, así como la secuencia nucleotídica de cada uno de los genes (tabla 1).
Tabla 1. Propiedades bioquímicas y moleculares de las proteínas de la membrana eritrocitaria
]]>
Las interacciones proteicas determinantes de la integridad de la membrana eritrocitaria son de 2 tipos: verticales y horizontales.2-4
Interacciones verticales®fijan el esqueleto a la doble capa lipídica.
Interacciones horizontales® son responsables de la estabilidad global del esqueleto.
Defectos en algunas de estas proteínas pueden dar lugar a trastornos clínicos en los cuales está involucrada la estabilidad del glóbulo rojo. Entre ellos los más frecuentes y mejor estudiados son la esferocitosis hereditaria (HS) y la eliptocitosis hereditaria (HE).4,7,8
La esferocitosis hereditaria (EH) es una enfermedad caracterizada por anemia hemolítica de severidad variable, con presencia de esferocitos en sangre periférica y una respuesta clínica favorable a la esplenectomía.15
Fue descrita en 1871 por 2 médicos belgas, Vanlair y Masius, cuando estudiaron a una paciente joven que presentaba dolor abdominal, esplenomegalia asociada con íctero, vómitos, anemia y marcada atrofia muscular. Los investigadores notaron que los glóbulos rojos tenían una forma esférica y mucho más pequeña que los normales, demostraron que el bazo estaba involucrado en el envejecimiento de estas células y denominaron a esta enfermedad microcitemia.16
Veinte años después fue redescubierta por Wilson y Minkowsky, quienes registraron 8 casos en 3 generaciones diferentes de una misma familia.17
La mayor contribución al conocimiento de esta enfermedad fue hecha por los hallazgos de Chauffard, el cual confirmó el aumento de la fragilidad osmótica de los eritrocitos, lo que explicaba la anemia hemolítica encontrada en estos casos. Observó también cómo se producía la corrección de la hemólisis con la esplenectomía, y demostró la implicación del bazo en esta entidad.18 Posteriormente se encontró que los glóbulos rojos de pacientes con HS presentaban una disminución de Na+ intracelular y una pérdida de lípidos de la membrana, lo que explicaba la disminución del área superfical de la célula.19 Por estos estudios la entidad se conoce también como enfermedad de Minkowsky-Chauffard.3-4
Después de la década de los 70, con el desarrollo de nuevas técnicas, se encontraron las primeras alteraciones bioquímicas de las proteínas de la membrana eritrocitaria y a partir de 1985, por medio de las técnicas del ADN recombinante, se han podido precisar las alteraciones moleculares en un número importante de casos.20
La EH es la anemia hemolítica más frecuente en el mundo y se ha señalado una prevalencia de 1:2000 en algunos países europeos.8 Sin embargo, estos datos pueden no reflejar la frecuencia real de la enfermedad, ya que no se tienen en cuenta los portadores asintomáticos ni los hallazgos de una frecuencia del 1 % de donantes de sangre con fragilidad osmótica aumentada observada en algunos países.15 Aunque es más frecuente en individuos de la raza blanca, puede observarse ocasionalmente en otras razas o grupos étnicos.15,21
El 75 % de las familias afectadas muestran un patrón autosómico dominante. El homocigótico de esta forma de herencia no ha sido identificado, lo que sugiere que sea incompatible con la vida.22 El 25 % restante corresponde a un patrón autosómico recesivo, nuevas mutaciones o pacientes con EH dominante con penetrancia incompleta.2,23
La EH es una enfermedad muy heterogénea desde el punto de vista clínico. Se puede observar desde el portador asintomático hasta pacientes que presentan una anemia hemolítica crónica con grandes requerimientos transfusionales.24,25
]]> Dependiendo de la severidad del cuadro clínico, de las cifras de hemoglobina, los niveles de bilirrubina y el conteo de reticulocitos, esta enfermedad se clasifica en 4 formas: portador asintomático, EH ligera, EH típica y EH severa.26,27Portador asintomático. En algunas familias se ha señalado un patrón de herencia autosómico recesivo. En estos casos, los padres de un paciente afectado no presentan ninguna alteración. En ocasiones la afectación es muy leve, como ligero incremento de las cifras de reticulocitos, escasos esferocitos en periferia o fragilidad osmótica incubada alterada y puede no ser detectada por los exámenes de rutina. Debe tenerse en cuenta también que pueden ocurrir nuevas mutaciones dentro de una familia aparentando un patrón de herencia autosómico recesivo, por lo que siempre es importante un estudio minucioso de todos los miembros de la familia.3,4
EH ligera. Comprende entre el 20 y 30 % de todos los pacientes con EH autosómica dominante, los que pueden presentar una hemólisis ligera compensada.3,27 Los individuos son frecuentemente asintomáticos y algunos casos son difíciles de diagnosticar, ya que la anemia y la esplenomegalia son muy ligeras y en ocasiones pueden estar ausentes.28 Muchos de estos pacientes se diagnostican durante estudios familiares o cuando en la etapa adulta aparece el íctero y la esplenomegalia. Episodios hemolíticos pueden presentarse en el curso de algunos procesos infecciosos como mononucleosis, parvovirus o citomegalovirus, así como durante el embarazo, por esfuerzos físicos intensos o por sangramientos.27-30
EH típica. Entre el 50 y 60 % de los pacientes con EH autosómica dominante tienen esta forma clínica. Presentan una hemólisis compensada incompleta y una anemia de ligera a moderada. El íctero es común en niños, aunque se puede ver también en los adultos y está asociado con infecciones virales ligeras, debido a la estimulación reticuloendotelial y a un aumento de la hemólisis. Los requerimientos transfusionales son esporádicos. La esplenomegalia está presente en el 50 % de los niños y en el 75 % de los adultos.2,3,31,32
EH severa. Estos pacientes (5-10 %) evolucionan con una hemólisis severa y presentan frecuentes requerimientos transfusionales. La mayoría de estos casos tienen una forma autosómica recesiva de la enfermedad. Pueden presentar crisis aplásticas, retardo del crecimiento y de la maduración sexual. La esplenectomía es el tratamiento de elección en esta forma clínica.33,34 Generalmente la enfermedad debuta al nacimiento con ictericia y hemólisis y se requiere, en muchas ocasiones, de exanguinotransfusión.30,32
Es bien conocido que la EH se produce por un defecto intrínseco del glóbulo rojo y se han demostrado defectos moleculares de diferentes proteínas que conforman el esqueleto de la membrana eritrocitaria. La presencia de alteraciones del metabolismo, del transporte catiónico, de la fosforilación de las proteínas y de la composición de los lípidos de la membrana, son secundarias a la causa primaria de esta enfermedad.3,8,15
La lesión en la membrana está dada por una pérdida del área de la célula, pero se desconoce si esto se debe a una pérdida física (fragmentación ) o a una contracción de la superficie de la membrana, y aunque la mayoría de las evidencias favorecen la fragmentación, no parece ser el único mecanismo que explique este hallazgo. Se ha demostrado que la fuerza requerida para fragmentar las células esferocíticas, así como su elasticidad y deformabilidad, están disminuidas, y que ésta es proporcional a la densidad de la espectrina en la membrana. Además, los glóbulos rojos esferocíticos pierden membrana más rápidamente que los normales cuando se deprime su metabolismo. La concentración de fosfolípidos y colesterol es del 15 al 20 % menor de lo normal debido posiblemente a la disminución de su superficie y se presume que también se produce una pérdida de las proteínas integrales de la membrana.2,4,25,31,35
El problema fundamental de la EH es la consecuencia reológica de la disminución de la relación superficie/volumen. La membrana del glóbulo rojo es muy flexible, pero sólo puede incrementar su área un 3 % antes de romperse. Por consiguiente, mientras la célula se vuelve más esférica, es cada vez menos deformable. En el caso de los hematíes esferocíticos, esta pobre deformabilidad es un obstáculo sólo para el bazo, ya que la mayoría de los esferocitos sobreviven bien después de la esplenectomía.4
- Eritrostasis:
Ham, Castle y Dacie fueron los primeros en señalar que los glóbulos rojos esferocíticos son particularmente vulnerables a la eritrostasis.
]]> El glóbulo rojo sufre una serie de cambios que conlleva a la autohemólisis cuando se incuba en ausencia de glucosa, proceso que se acelera en la HS, lo que conlleva a que los esferocitos se depleten de ATP más rápidamente que lo normal. A medida que los niveles de ATP disminuyen, falla la bomba de cationes y penetra agua y sodio en la célula. Cuando la célula alcanza muy bajos niveles de ATP, el calcio intracelular también aumenta, y da lugar a un fallo en la bomba de calcio, lo que produce una salida del potasio intracelular. El mecanismo molecular de estos cambios en la permeabilidad no es bien conocido, pero sus consecuencias están bien definidas. A medida que el potasio disminuye, el agua responde al cambio en osmolaridad y las células se encogen. Los hematíes esferocíticos no son capaces de soportar estos cambios, ya que son inestables y se fragmentan excesivamente durante la depleción metabólica, los lípidos de la membrana se pierden a una velocidad superior al doble de la normal y aunque no se sabe con exactitud, se plantea que también existe una pérdida proporcional de proteínas integrales de la membrana (al menos en la deficiencia primaria de banda 3 se ha demostrado). Aunque la disminución de la superficie inicialmente se acompaña de una deshidratación celular, la pérdida de membrana predomina, la célula excede su volumen de hemólisis crítico (volumen/superficie > 100) y se produce la autohemólisis.1-4- Dinámica del atrapamiento esplénico.
Uno de los aspectos no conocidos acerca de la fisiopatología de la EH es si los eventos que dan lugar al acondicionamiento y destrucción de los glóbulos rojos esferocíticos en el bazo son los mismos que dan lugar a un incremento en la esferoidicidad y autohemólisis durante la eritrostasis in vitro.
Es conocido que los hematíes esferocíticos son selectivamente retenidos por el bazo, y producen una pérdida de la membrana, lo que promueve el atrapamiento esplénico y la destrucción eventual de la célula. Estudios han demostrado que el tiempo de tránsito esplénico medio se correlaciona inversamente con la supervivencia de los glóbulos rojos en la HS. Parece ser que el atrapamiento esplénico es promovido inicialmente por la inestabilidad del esqueleto de la membrana, pero los detalles de cómo el defecto molecular permite que éste se produzca, aún no se han definido. Los mecanismos del condicionamiento esplénico y de la destrucción eritrocitaria son todavía desconocidos. Estudios cinéticos sugieren que los glóbulos rojos son atrapados continuamente dentro de los cordones esplénicos durante el período que se requiere para inducir la forma esferoidal pasiva y la autohemólisis por depleción metabólica, aunque otra posibilidad puede ser la presencia de daños metabólicos repetidos. Una especial susceptibilidad de los hematíes esferocíticos al medio ácido del bazo y una intervención activa de los macrófagos en el proceso de daño celular durante la eritrostasis, pueden influir también, pero no hay evidencias directas sobre estas 2 hipótesis (fig.3).2,4,15,23,31,35

Fig. 3. Fisiopatología del condicionamiento esplénico y destrucción de los glóbulos rojos en la esferocitosis hereditaria (EH).
Los hallazgos morfológicos encontrados en la EH son los mismos que encontramos en otros procesos hemolíticos: hiperplasia de precursores eritroides en la médula ósea, cifra elevada de reticulocitos en sangre periférica, aumento de la bilirrubina no conjugada en el plasma y una elevada excreción de urobilinógeno.
La anemia generalmente es normocítica, normocrómica, con volumen corpuscular medio (VCM) normal o subnormal, aunque menor del esperado en otras condiciones con un grado similar de reticulocitosis.
La hemoglobina corpuscular media (HCM) es normal, pero la concentración hemoglobínica corpuscular media (CHCM) está aumentada en el 50 % de los pacientes, lo que refleja una deshidratación de una parte de la población celular. No obstante, lo que caracteriza a la EH es la presencia de esferocitos en sangre periférica.2-4
Las diversas formas de la EH muestran un patrón morfológico diferente entre cada una de estas. Los pacientes con EH autosómica dominante y algunos con la forma autosómica recesiva muestran solamente esferocitos en la lámina de periferia, mientras que pacientes con una deficiencia severa de b espectrina, presentan acantocitos y poiquilocitos, pudiendo en algunos casos, aparecer estos últimos en un mayor porcentaje que los esferocitos. En los casos con una deficiencia de banda 3 se ha observado también la presencia de pincered cells.3,4,8,31,35
]]> La prueba más utilizada para el diagnóstico de la EH es la fragilidad osmótica del glóbulo rojo, la cual mide la habilidad de los glóbulos rojos de incrementar su volumen cuando son sometidos a soluciones hipotónicas de cloruro de sodio (NaCl) de concentraciones variables. Debido a que los esferocitos tienen una relación superficie/volumen disminuida, tienen una capacidad disminuida para aumentar su volumen y se lisan a una concentración de sales más elevada que las células normales. La sensibilidad de esta prueba puede incrementarse con la preincubación de las células.31 Se han señalado otras modificaciones de la prueba de fragilidad osmótica. Entre estas está la autohemólisis, la cual determina la hemólisis de los glóbulos rojos incubados sin glucosa en condiciones estériles, la prueba del glicerol y la prueba rosa,31 pero ninguna de ellas parece ser más sensible que la fragilidad osmótica incubada. Se ha señalado que la prueba de criohemólisis hipertónica es 100 % sensible para el diagnóstico de la EH, pero estos resultados no han sido debidamente confirmados todavía.36 La introducción de la ectacitometría, que cuantifica la deformabilidad de los eritrocitos por la medición de la fuerza que induce la elongación de la célula, permitió el desarrollo de la ectacitometría osmótica, la cual parece ser la prueba más sensible en la actualidad.37 Sin embargo, esta técnica solo está disponible en un reducido grupo de laboratorios especializados. La fijación con glutaraldehído es útil para detectar la presencia de alteraciones morfológicas: microcitos, acantocitos, pincered cells, poiquilocitos, etcétera.37El análisis de las proteínas de la membrana eritrocitaria en electroforesis de poliacrilamida con SDS (PAGE-SDS) se utiliza para cuantificar las proteínas y/o detectar péptidos truncados con migración anormal, y el estudio molecular ha permitido identificar un número importante de mutaciones en distintas proteínas de la membrana eritrocitaria (fig.4).
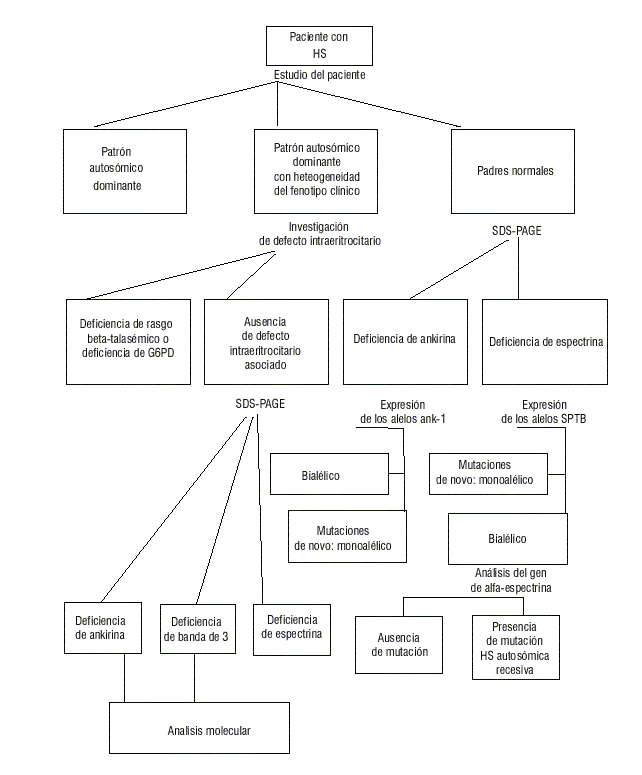
Fig.4. Algoritmo para el estudio de un paciente con esferocitosis hereditaria (EH).
Es necesario señalar que para un diagnóstico preciso de la EH es importante siempre realizar el estudio familiar.
La forma típica de EH puede ser diagnosticada fácilmente, aunque deben descartarse otras causas de anemia hemolítica esferocítica tales como la anemia hemolítica autoinmune (prueba de Coombs), hemoglobinas inestables (electroforesis de hemoglobina, cuerpos de Heinz, etc.), la estomatosis hereditaria y el síndrome del Rh nulo, entre otras. Durante el período neonatal, es difícil diferenciar la EH de la incompatibilidad ABO, en la que la esferocitosis es evidente. En estos casos, es imprescindible el estudio familiar, así como una reevaluación del niño entre los 4 y 6 meses de nacido. No obstante, los esferocitos de la EH son distinguibles de las anemias hemolíticas autoinmunes por la uniformidad de éstos, así como por el aumento de la CHCM.1-4,8
Se presentan también dificultades diagnósticas en los pacientes que debutan con una crisis aplástica. Al inicio, la naturaleza de los síntomas sugiere la aparición de un proceso adquirido y la ausencia de reticulocitos puede enmascarar el diagnóstico de la anemia hemolítica. También se puede confundir el diagnóstico cuando la EH se asocia con otras enfermedades que elevan la relación volumen/superficie, tales como la deficiencia de hierro o el íctero obstructivo. La deficiencia de hierro normaliza la fragilidad anormal y la forma de los esferocitos, pero no mejora la sobrevida de éstos. En el íctero obstructivo desaparece la esferocitosis, debido a la acumulación de colesterol y fosfolípidos en la membrana celular. En individuos normales, este proceso conlleva a la formación de target cells, mientras que en la HS las células adoptan una forma discoidal y se incrementa la sobrevida de las células. La b-talasemia heterocigótica y algunas enzimopatías, también pueden interferir en el diagnóstico de la HS.15,23,31
Debido al curso asintomático de esta enfermedad en muchos pacientes, debe descartarse la presencia de una EH en aquellos casos con síntomas aislados tales como esplenomegalia, íctero, litiasis vesicular en el adulto joven, anemias como resultado de mononucleosis infecciosa u otras infecciones severas y durante el embarazo.1,8,15,35
Ocasionalmente se pueden observar esferocitos en pacientes con marcada esplenomegalia (cirrosis y mielofibrosis) o en pacientes con anemias microangiopáticas, pero el diagnóstico diferencial de estas entidades no presenta grandes dificultades.15
El estudio familiar es muy importante en el diagnóstico diferencial, sobre todo para poder precisar el carácter hereditario de la anemia, ya que las pruebas empleadas para el diagnóstico de la HS pueden ser positivas en muchas de las patologías mencionadas anteriormente.2-4,8
La crisis aplástica debida a la infección por el parvovirus B19, es la complicación más importante que pueden presentar estos pacientes, la cual puede acompañarse de fiebre, vómitos, dolor abdominal, cefalea, palidez y anemia severa.15,23,31
Las crisis megaloblásticas resultan de una ingestión insuficiente de ácido fólico. Se observan frecuentemente durante el embarazo, donde los requerimientos de ácido fólico son más elevados.4
La litiasis vesicular fue la primera complicación descrita en los pacientes con EH, la cual se ha observado en niños pequeños, aunque su aparición es más frecuente en adolescentes y adultos. Entre el 55 y el 85 % de pacientes con EH presentan litiasis, y la mitad de estos tienen síntomas de colelitiasis o de obstrucción biliar.2-15
Otras complicaciones, aunque muy poco frecuentes, han sido señaladas: hematopoyesis extramedular, gota, úlceras en miembros inferiores y retraso en el crecimiento y en el desarrollo sexual.26,28,31,35
La esplenectomía es el tratamiento de elección para los pacientes con EH, ya que es el más efectivo en el control de la anemia, aunque la sobrevida de los glóbulos rojos permanece acortada y los esferocitos no desaparecen. Está indicada en pacientes con anemia hemolítica severa o en individuos moderadamente sintomáticos, pero que presentan litiasis vesicular. En el resto de los casos con EH existe discrepancia en cuanto la recomendación de realizar la esplenectomía. Algunos investigadores sugieren realizarla a todo paciente con HS, aún en aquéllos con una forma ligera o moderada de la enfermedad y en ausencia de litiasis vesicular, mientras que otros plantean un manejo más conservador. La decisión de la esplenectomía en tales pacientes debe ser analizada individualmente, evaluando los riesgos y beneficios en cada caso. En los niños, este proceder debe ser demorado lo más posible y no se recomienda su indicación antes de los 5 años de edad, ya que el riesgo de infección es muy elevado.8,15,18,33
En la forma típica de la enfermedad, después de la esplenectomía, la esferocitosis persiste, pero el condicionamiento de los microesferocitos desaparece, los niveles de reticulocitos se acercan a la normalidad y aumenta el tiempo de sobrevida de estos hematíes.8,15,31
Diferentes estudios han demostrado que el grado de respuesta a la esplenectomía está directamente relacionado con el grado de deficiencia de espectrina, sobre todo en la forma severa de la enfermedad (fundamentalmente la forma autosómica recesiva), por lo que los pacientes mejoran el cuadro clínico y hematológico, desaparecen los requerimientos transfusionales, pero en muchos casos, se mantiene una hemólisis de intensidad variable.26
La complicación más severa de este tratamiento es la sepsis posesplenectomía. Los datos reportados en la literatura indican que esto ocurre en el 3,5 % de los pacientes con EH, y de éstos, en el 60 % es fatal. Teniendo en cuenta el riesgo de infección, estos pacientes se inmunizan con distintas vacunas: neumococo polivalente, Haemophilus influenzae y antimeningococo. Resultados satisfactorios se han obtenido también con la terapia antibiótica profiláctica con Fenoximetilpenicilina durante 5 años después de la operación. Existen pacientes que no responden a la esplenectomía, lo cual puede deberse a: 1. La presencia de un bazo accesorio. 2. El desarrollo de esplenosis, la cual puede ocurrir varios años después de la esplenectomía. 3. La presencia de otra causa de anemia hemolítica concomitante con la EH.4,15,31
Debido al riesgo inherente de las infecciones posesplenectomía, se han empleado en los últimos años métodos más conservadores:
El análisis de las proteínas de la membrana mediante electroforesis en gel de poliacrilamida (SDS-PAGE), ha sido un elemento muy importante para el estudio de las alteraciones de las proteínas del glóbulo rojo en la HS. Empleando un sistema de buffer continuo de Fairbanks, con un gradiente exponencial de poliacrilamida de 3,5 al 17 %, se puede identificar la presencia de péptidos truncados o elongados, o una concentración anormal de las proteínas que conforman el citoesqueleto del glóbulo rojo.3,8,41
Alteraciones de 4 proteínas de la membrana eritrocitaria han sido identificadas en la EH y se ha demostrado que esta enfermedad se produce por defectos de las proteínas que intervienen en las interacciones verticales entre el esqueleto de la membrana y la bicapa lipídica,4,8,31,42-44 que son:
Diferentes estudios bioquímicos han señalado que del 30 al 45 % de los pacientes tienen deficiencia combinada de Sp y ankirina, el 30 % presenta deficiencia de Sp aislada y el 20 % muestra deficiencia de banda 3. Sin embargo, se plantea que el porcentaje de individuos con deficiencia de ankirina está subestimado por problemas de sensibilidad de la técnica, y además porque la reticulocitosis enmascara también la deficiencia de esta proteína. Se ha demostrado que cuando existe una deficiencia de ankirina, la espectrina que está en exceso es incapaz de ensamblarse a la membrana, debido a la pérdida de los sitios de unión de la ankirina, y por este motivo es degradada. Esta es la razón por la que frecuentemente se produce una deficiencia de ambas proteínas; la deficiencia de espectrina es secundaria a una reducción primaria de ankirina. El defecto aislado de proteína 4.2 se ha encontrado en un reducido número de americanos y europeos, pero es más común en Asia, especialmente en Japón. Esta variedad de fenotipos bioquímicos refleja la amplia heterogeneidad de las alteraciones moleculares que se producen en la EH.45-48
Al nivel molecular, las mutaciones que dan lugar a las alteraciones señaladas anteriormente se encuentran distribuidas a lo largo de los genes afectados, y pueden ser debidas a (tabla 2. fig.5):
Tabla 2. Alteraciones moleculares encontradas en la esferocitosis hereditaria (EH)
| ]]> Proteína | Mutaciones que producen corrimiento del marco de lectura | Mutaciones sin sentido (codon de terminación) | Mutaciones puntiformes | Mutaciones en el sitio de empalme | ]]>
Deleciones grandes del genoma | Total | |
| Deleciones | Inserciones | ||||||
| Espectrina ]]> | |||||||
| a Sp | 1 | - | ]]> 1 | 2 | 1 | - | 5 |
| b Sp | 3 | 2 | 2 | 1 | - | ]]> 1 | 9 |
| Ankirina | 7 | 2 | 2 | 7 | - | - | 18 |
| Banda 3 | 6 | ]]> 4 | 2 | 12 | - | - | 24 |
| Proteína 4.2 | 2 | - | 1 | 2 | ]]> - | - | 5 |
| Total | 19 | 8 | 8 | 24 | 1 | 1 | 61 |
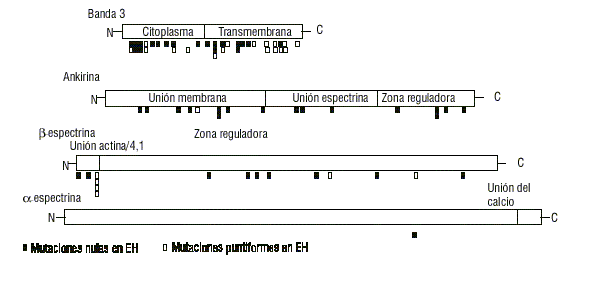
Fig. 5. Esquema de los dominios de banda 3, ankirina, a y b espectrina con la ubicación de las mutaciones conocidas que causan esferocitosis hereditaria (EH).
Estos defectos moleculares pueden afectar la estabilidad de la transcripción, la estabilidad de la proteína mutada o la función específica de la proteína. Todas estas consecuencias conllevan a una deficiencia de la proteína alterada. En el caso de las mutaciones puntiformes, no se conoce el mecanismo por el cual se produce un defecto de la proteína. Sin embargo, algunas de estas mutaciones se encuentran en residuos de aminoácidos altamente conservados, que dan lugar a una proteína con un daño funcional significativo. Es importante señalar que, mientras la expresión del gen de la a espectrina está limitada a los eritrocitos, los genes de la b espectrina, ankirina, banda 3 y proteína 4.2 se expresan en otras formas alternativas en otros tejidos, por lo que algunas mutaciones de estos genes se han reportado en asociación con manifestaciones extraeritrocitarias.2-4,6,13,15,21-23,31,35,49,50
Los genes de la espectrina, ankirina y banda 3 contienen o están flanqueados por polimorfismos conocidos, que son útiles para estudios de ligamiento y consisten en mutaciones puntiformes, número variable de tandem repetidos (NVTR) o número variable de dinucleótidos repetidos (NVDR) (tabla 3).51,52
Tabla 3. Algunos polimorfismos silentes de la espectrina y la ankirina

Deficiencia de espectrina:
Tabla 4. Algunos polimorfismos silentes de la proteína banda 3
| Nombre | Cambio | Exon | Intron | Consecuencia | Dominio |
| Genas | 89G®A | ]]> 1 | - | síntesis | 3´UT |
| Pst I | - | - | 3 | - | CP |
| ´DA38A´ | 227A®C | ]]> 4 | - | GAC®GCC; D38A | CP |
| Memphis I | 280A®C | - | - | AGG®GAG; K56E | CP |
| Napoli | 411 ins T | ]]> 5 | - | Corrimiento de lectura | CP |
| Nachod (Hadreckralove II) | 464(-3c®a) | 5 | - | Deleción | CP |
| Mondego | 553C®T | ]]> 6 | - | CTT®TCT; P147S | CP |
| Memphis III | 2675C®T | 19 | - | CCG®CTG; P854L | TM13 |
CP:citoplasmático; TM: transmembranoso.
]]> Deficiencia de ankirina:Diferentes investigadores han señalado que la deficiencia de ankirina está presente en un número importante de pacientes con HS. Puede ser debido a translocaciones o a delaciones del brazo corto del cromosoma 8, aunque se han encontrado también mutaciones puntiformes, sin sentido y en el sitio de empalme. Se ha demostrado que en la tercera parte de los pacientes con deficiencia combinada de ankirina y espectrina, uno de los alelos de ankirina tiene una expresión reducida. Esto puede ser debido a una reducción en la transcripción del gen o a una disminución de la estabilidad de los transcriptos. También se ha observado que las mutaciones de novo en uno de los alelos de la ankirina, que dan lugar a una disminución en su expresión, se encuentran con cierta frecuencia en los pacientes con HS cuyos padres son normales.44,53
Deficiencia de banda 3:
La deficiencia de banda 3 en pacientes con EH se ha demostrado mediante la electroforesis en gel de poliacrilamida (PAGE-SDS) y se plantea que está presente entre el 10 y el 20 % de los casos con la forma dominante. En ocasiones, puede verse acompañada de una disminución de la proteína 4.2 como consecuencia de mutaciones que afectan la porción citoplasmática de la banda 3. Muchos pacientes tienen anemia ligera, esferocitosis y del 0,2 al 2,3 % de mushroom-shaped o pincered cells, lo cual no se ha encontrado en otras formas de HS.8,54-56
Deficiencia de banda 4.2:
Se ha demostrado la disminución severa o ausencia de la proteína 4.2 en la forma recesiva de la EH. Los fenotipos descritos en la literatura son muy heterogéneos: la morfología celular ha sido caracterizada por la presencia de esferocitos, eliptocitos o esferoovalocitos. Se ha encontrado fundamentalmente en Japón.57,58 La variabilidad del cuadro hematológico, bioquímico y clínico de la EH dentro de una misma familia, ha hecho pensar en la presencia de otros factores intrínsecos o mutaciones silentes que modulen la expresión de los alelos patológicos. Recientemente se han presentado algunos hallazgos que pudieran explicar el comportamiento tan heterogéneo de esta enfermedad.59,60
Futuras investigaciones permitirán profundizar en la relación entre las alteraciones moleculares y la expresión clínica de la EH.
Subject headings: SPHEROCYTOSIS, HEREDITARY/diagnosis; SPHEROCYTOSIS, HEREDITARY/genetics; DNA RECOMBINANT; MEMBRANE PROTEINS/chemistry; RECOMBINANT PROTEINS/chemistry; SPLENECTOMY; ERYTHROCYTE MEMBRANE/chemistry.
Recibido: 5 de abril del 2001. Aprobado: 21 de diciembre del 2001.
Lic. Mayelín Herrera García. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800, Ciudad de La Habana, Cuba. Telef.(537)578268. Fax (537)338979.e-mail: m.herrera@hemato.sld.cu