ARTÍCULO ORIGINAL
Estudio hematológico, bioquímico y clínico de pacientes y familiares con esferocitosis hereditaria
Hematologic, biochemical and clinical study of patients and relatives presenting with inheritable spherocytosis
DraC. Marianela Estrada del Cueto; MsC. Mayelín Herrera García; Dra. Clara Mayo de las Casas; Dra. Tamy Robaina Herrera; Dra. Valia Pavón Morán; Dr. Juan Carlos Jaime Facundo; Dr. Antonio Bencomo Hernández; Téc. Graciela Pérez Diez de los Ríos; Téc. Ana Hernández Martínez ]]>
Instituto de Hematología e Inmunología. Ciudad de La Habana, Cuba.
Se realizó un estudio hematológico, bioquímico y clínico a 6 pacientes diagnosticados con esferocitosis hereditaria (HS) y a sus familiares (40). Se encontraron 13 familiares portadores de la enfermedad. En 4 familias, el patrón de herencia fue autosómico dominante y en 2 de ellas no se pudo precisar. Los 19 casos se clasificaron en: 6 (31,6 %) portadores asintomáticos, 2 (10,5 %) con esferocitosis hereditaria ligera, 9 (47,4 %) con la forma típica y 2 (10,5 %) con la forma severa. Diez (52,6 %) tenían defecto de espectrina, de ellos, 6 (31,5 %) con otra alteración asociada. En 8 (42,1 %) no se pudo precisar el defecto bioquímico. No observamos relación entre la alteración bioquímica y el cuadro clínico de la enfermedad. La expresión hematológica, bioquímica y clínica fue muy heterogénea entre las diferentes familias y entre los miembros afectados de cada una de ellas. La alteración bioquímica más frecuente fue la deficiencia de espectrina. Nuestros resultados son similares a los señalados por otros investigadores.
Palabras clave: esferocitosis hereditaria, anemia hemolítica, proteínas de la membrana eritrocitaria.
A hematologic, biochemical and clinical study was conducted in 6 patients diagnosed with inheritable spherocytosis (IS) and its relatives (40). There were 40 family carriers of this disease. In 4 family, the inheritance pattern was autosomal dominant, and in two of them it was impossible its determination. The 19 cases were classified in: 6 (31,6%) asymptomatic carriers,2 (10,5%) with light-inheritable spherocytosis, 9 (47,4%) con the typical presentation, and 2 (10,5%) with the severe presentation. Ten (52,6%) had spectrine defect , from them, 6 (31,5%) with another associated alteration. In 8 (42,1%) it was impossible to determine the biochemical defect. There was not relation between biochemical alteration and the clinical picture of this disease. Hematologic, biochemical and clinical expression was very heterogeneous among the different families and among the members affected of each. The more frequent biochemical alteration was the spectrine deficiency. Our results are similar to reported by other researchers.
]]> Key words: Inheritable spherocytosis, hemolytic anemia, erythrocyte membrane proteins.
INTRODUCCIÓN
La esferocitosis hereditaria (HS) es una enfermedad producida por un trastorno de las proteínas que conforman el citoesqueleto de la membrana eritrocitaria. Aunque está presente en todos los grupos raciales, es particularmente común en individuos del norte de Europa con una prevalencia de aproximadamente 1:3000.1,2 El patrón de herencia más frecuente es el autosómico dominante (75 %). El resto de los casos presentan un patrón autosómico recesivo, aunque excepcionalmente se han estudiado pacientes con historia familiar negativa y mutaciones de novo.3,4
Los genes responsables de la HS incluyen: ankirina (proteína 2.1), b espectrina (Sp), proteína banda 3, a Sp y proteína 4.2. 3 En la HS típica, el defecto más frecuente es la presencia de mutaciones de ankirina, seguido por la deficiencia de banda 3 y de b Sp. En los casos con la forma recesiva de la enfermedad predominan los defectos de a espectrina o de la proteína 4.2.5-7.
Las manifestaciones clínicas de la HS son muy variadas. En la HS típica hay evidencia de hemólisis con anemia, ictericia, reticulocitosis, litiasis, esplenomegalia, esferocitos con reducción del área de la membrana celular, resistencia osmótica disminuida y generalmente existe una historia familiar positiva. La mayoría de los casos tienen una hemólisis compensada incompleta y anemia ligera o moderada. En ocasiones, la anemia es difícil de detectar. La ictericia se observa en la mitad de los individuos y aparece asociada con infecciones virales. La esplenomegalia se puede detectar en los niños mayores o en los adultos; el 25 % de los pacientes tienen hemólisis compensada y la anemia es ligera o está ausente. En un número reducido de casos, la hemólisis no está compensada y la anemia es severa, lo que se observa fundamentalmente en la forma recesiva de la enfermedad. Estos pacientes generalmente son dependientes de transfusiones y pueden desarrollar algunas complicaciones como retraso en el crecimiento y en la maduración sexual, tumores extramedulares, úlceras maleolares o facies talasémica.4,5,8
Entre las complicaciones clínicas que pueden presentar está la esplenomegalia, la litiasis vesicular y la obstrucción biliar. Como se observa también en otros tipos de anemia hemolítica congénita, la asociación de la HS con el síndrome de Gilbert incrementa el riesgo de ictericia neonatal y de litiasis vesicular.9,10
Para diagnosticar a un paciente con HS es indispensable el estudio familiar. Los estudios moleculares han demostrado que los hallazgos morfológicos se asocian con ciertos defectos de las proteínas de la membrana: pincered eritrocitos (banda 3), acantocitos esferocíticos (b Sp) o esferoestomatocitos (proteína 4.2). 4-6 La concentración hemoglobínica corpuscular media (CHCM) se encuentra aumentada entre el 35 y 38 % de los casos y en el 50 % de estos es debido a una deshidratación celular. La resistencia osmótica y la prueba del glicerol están disminuidas, y otros marcadores de hemólisis, como los reticulocitos, la bilirrubina indirecta, la deshidrogenasa láctica y el urobulinógeno fecal, y en orina, se encuentran aumentados, mientas que la haptoglobina está disminuida, lo que refleja un incremento en la producción o destrucción de los eritrocitos. El análisis de las proteínas de la membrana eritrocitaria mediante electroforesis en gel de poliacrilamida (sodio dodecil sulfato, SDS-PAGE), ha sido un elemento muy importante para el estudio de las distintas alteraciones bioquímicas, tales como la presencia de péptidos truncados o alongados, o una concentración anormal de las proteínas que conforman el citoesqueleto del glóbulo rojo.4,11,12 ]]>
La esplenectomía total mejora el cuadro de anemia en la mayoría de los pacientes con HS, pero los riesgos de infección y muerte son elevados. Desde la década de los 90 del siglo pasado, se ha comenzado a realizar la esplenectomía parcial con resultados satisfactorios en cuanto a la recuperación hematológica de los pacientes con hemólisis severa y con la ventaja de que generalmente no presentan complicaciones posquirúrgicas. 13En este trabajo nos proponemos realizar la evaluación y caracterización hematológica, bioquímica y clínica de 6 pacientes diagnosticados con HS y de sus familiares.
MÉTODOS
Se estudiaron 6 niños con HS diagnosticados en el Instituto de Hematología e Inmunología. Se confeccionó el árbol genealógico de cada uno de ellos (4 generaciones) y se evaluaron también los familiares disponibles (41 individuos).
Descripción de los casos
]]>
Caso 1. RVT: Paciente del sexo femenino, raza blanca, de 5 años de edad, que fue remitida a nuestro centro por la consulta de Puericultura a los 2 meses y medio de edad por presentar palidez y hemoglobina (Hb) en 8 g/dL. Se realizó el dignóstico de HS. Las cifras de Hb oscilaron entre 8, 4 y 12,2 g/dL y los reticulocitos entre 1,2 y 10,2 %. No ha tenido requerimientos transfusionales. Solo en una ocasión (al año de edad), se detectó visceromegalia. Hay antecedentes de la enfermedad por vía materna. Los familiares estudiados fueron: III-2 padre, III-3 madre, III-4 tío materno, II-4 abuela materna, II-5 abuelo paterno.
Caso 2. GCPM: Paciente del sexo femenino, raza blanca, de 6 años de edad, con enfermedad de Perthes, que fue estudiada en nuestro centro a solicitud de la madre, ya que existían antecedentes de esta enfermedad por vía materna. Fue diagnosticada como portadora de HS. Nunca se ha transfundido y no presenta hepatoesplenomegalia. Las cifras de Hb oscilaron entre 12,1 y 13,1 g/dL y los reticulocitos entre 1,2 y 5,4 %. Los familiares estudiados fueron: I-1 bisabuela materna, II-5 tía abuela materna, II-8 abuela materna, III-1 tío por vía materna, III-2 tío por vía materna, III-5 tío por vía materna, III-8 madre, III-9 padre, IV-1 primo por vía materna.
Caso 3. AGH: Paciente del sexo femenino, raza blanca, de 2 años de edad, que fue remitida a nuestro centro cuando tenía 1 año por presentar Hb 7,5 g/dL y reticulocitos 9,6 %. Se realizó el diagnóstico de HS. Se transfundió al año y medio de edad por caída de la Hb a 6,3g/dL con reticulocitos en 4,4 %. Mantiene esplenomegalia entre 3 y 6 cm. Hay antecedentes de la enfermedad por vía materna. Los familiares estudiados fueron: II-2 abuelo materno, II-3 abuela materna, III-2 tío por vía materna, III-3 madre, III-4 padre, IV-1 primo por vía materna.
Caso 4. SSR: Paciente del sexo masculino, raza blanca, de 3 años de edad, con antecedentes de anemia desde los 3 meses de nacido. Se realizó el diagnóstico de HS al año y medio de edad. Las cifras de Hb han oscilado entre 9,4 y 11,1 g/dL y los reticulocitos entre 0,8 y 2,9 %. No ha presentado visceromegalia ni ha tenido requerimientos transfusionales. Los familiares estudiados fueron: II-5 abuelo materno, III-2 madre, IV-2 hermano.
Caso 5. BRR: Paciente del sexo masculino, raza blanca, de 1 año de edad, al que se le realizaron 2 exanguinotransfusiones al nacimiento debido a conflicto ABO. A los 2 meses de edad se detectó caída de las cifras de Hb (5,6 g/dL) y reticulocitosis (7,8 %), ictericia y esplenomegalia de 4 cm, después de recibir tratamiento con antibióticos por una septicemia. Se decidió transfundir y a los 5 meses y medio de edad es remitido a nuestro centro donde se realiza el estudio completo de anemia hemolítica y se concluyó como portador de HS. A los 9 meses de edad fue transfundido nuevamente por presentar Hb 6,5 g/dL, reticulocitos en 8 %, palidez cutánea y hepatoesplenomegalia. Desde los 2 meses de edad la Hb ha oscilado entre 5,8 y 12,8 g/dL y los reticulocitos entre 1,2 y 11 %. Mantiene hepatoesplenomegalia. No hay antecedentes de anemia en ningún miembro de la familia. Los familiares estudiados fueron: II-2 abuelo materno, II-3 abuela materna, II-17 abuelo paterno, II-18 abuela paterna, III-2 tío por vía materna, III-4 tía por vía materna, III-6 tía por vía materna, III-7 madre, III-8 padre, III-10 tío por vía paterna, IV-6 medio hermano por vía paterna.
]]>
Caso 6. JAVS: Paciente del sexo masculino, raza blanca y 3 años de edad que fue remitido a nuestro centro a los 2 años de edad por presentar Hb en 5,0 g/dL, reticulocitos en 15 % y esplenomegalia de 4 cm. La Hb ha oscilado entre 5 y 8,2 g/dL y los reticulocitos entre 8,8 y 15 %. No ha tenido requerimientos transfusionales. Mantiene esplenomegalia desde el diagnóstico y ligera hepatomegalia. No hay antecedentes familiares de la enfermedad. Los familiares estudiados fueron: II-10 abuela materna, II-11 abuelo materno, III-2 padre, III-3 madre, III-4 tío por vía materna, IV-2 hermano.
A cada paciente y sus familiares se les realizaron los siguientes estudios: Hb, reticulocitos, lámina de periferia,14 resistencia osmótica cualitativa y cuantitativa,15 prueba de glicerol,16 fijación con glutaraldehído,17 prueba de Coombs directa e indirecta,18 actividad y electroforesis de glucosa 6 fosfato deshidrogenasa (G6PD),15 electroforesis de Hb, Hb fetal (Hb F) y Hb A2, 15 extracción de membrana eritrocitaria19 y electroforesis de proteínas de la membrana eritrocitaria en gradiente de poliacrilamida 3,517 % SDS.11 Para la coloración se utilizó azul de Comassie y los geles fueron secados en papel de celulosa para su conservación y posterior análisis. La extracción de membrana se realizó a partir de 5 mL de sangre total con heparina y las muestras de pacientes y familiares se procesaron siempre con un control. Para obtener las concentraciones de cada proteína en el gel se empleó el programa Molecular Analyst de la BioRad instalado en una computadora con escáner. Con estos datos se determinaron las siguientes relaciones: (a + b)/banda 3; ankirina/(a + b), 4.1/(a + b) y 4.2/(a + b). Los valores normales para estas relaciones (X; media ± 1DS; desviación estándar) se obtuvieron mediante el estudio de 13 donantes de sangre.
Se revisaron las historias clínicas de cada paciente para recoger todos los datos hematológicos, bioquímicos y clínicos de interés. En el caso de los familiares, se realizó un examen físico completo y un interrogatorio detallado.
Ninguno de los pacientes o familiares estudiados tenía una enfermedad congénita o adquirida que pudiera influir en los resultados de las pruebas de laboratorio o en el cuadro clínico de la enfermedad.
RESULTADOS
Todos los pacientes y familiares estudiados son de la raza blanca, de ellos, 3 del sexo femenino y 3 del masculino. ]]>
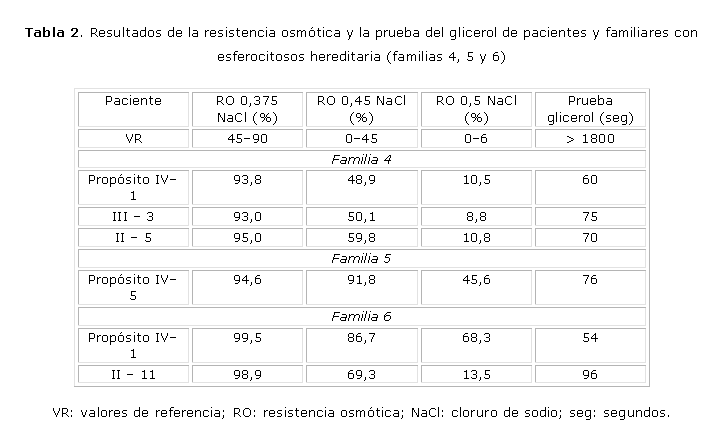
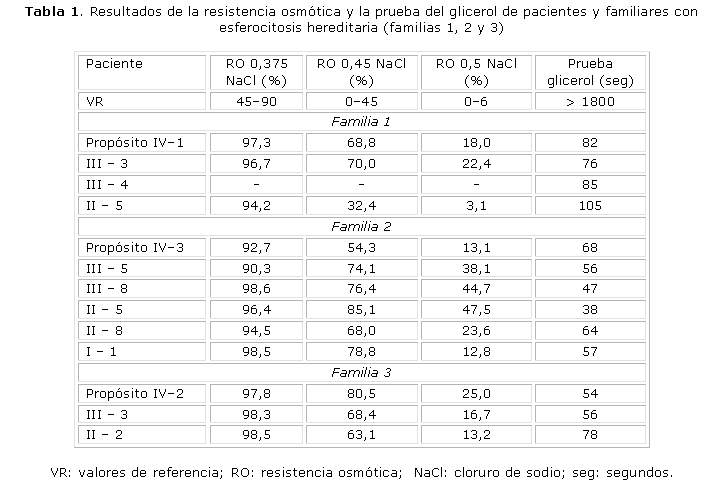
La electroforesis de Hb, la actividad y electroforesis de G6PD, la Hb F, Hb A2 y la prueba de Coombs directa e indirecta fueron normales en todos los pacientes y familiares estudiados. La FG y la lámina de periferia fueron normales, excepto en los pacientes con HS.En las tablas 1 y 2 se presentan los resultados de la resistencia osmótica y la prueba de glicerol de los 6 pacientes y familiares con HS. El total de familiares con HS fue: 3 en la familia #1, 5 en la familia #2, 2 en la familia #3, 2 en la familia #4, 1 en la familia #6 y ninguno en la familia #5 (13 en total). Todos los casos, excepto uno (familia 1; II-5), mostraron disminución de la resistencia osmótica en las 3 concentraciones de cloruro de sodio (NaCl). La prueba de glicerol estuvo disminuida en todos los pacientes y familiares con HS.
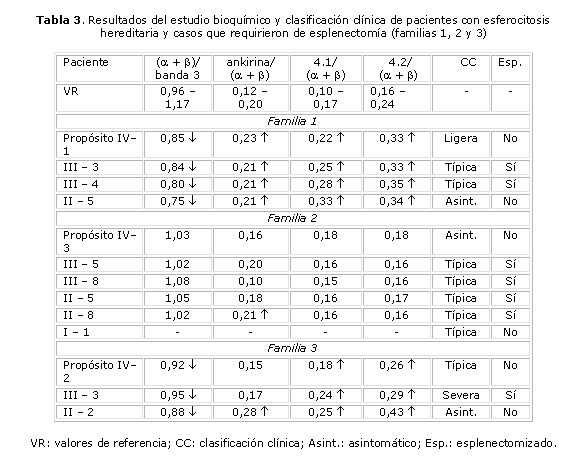
En las tablas 3, 4 y 5 se muestran los resultados del estudio bioquímico, la clasificación clínica de HS, así como los casos que requirieron de esplenectomía por causa de la enfermedad.
En la familia #1 se demostró un patrón de herencia autosómico dominante y los resultados bioquímicos permitieron concluir que la alteración bioquímica es producida por una deficiencia de Sp. Los resultados de los casos II-4 y III-2 fueron normales. El caso III-3 presentó ictericia neonatal con caída de las cifras de Hb a los 41 días de nacida. Mantenía niveles de Hb entre 8-9 g/dL y fue transfundida en 2 ocasiones. Se diagnosticó como portadora de HS a los 3 años y fue esplenectomizada a los 4 años de edad. Actualmente se encuentra asintomática. El caso III-4 presentó también ictericia neonatal, mantenía cifras de Hb alrededor de 9 g/dL y presentó varias crisis hemolíticas en los primeros meses de vida. Fue transfundido en 3 ocasiones. Se realizó el diagnóstico de la enfermedad a los 3 años y fue esplenectomizado a los 9 años de edad. Estos 2 casos fueron clasificados como HS típica. El propósito IV1 fue clasificado como HS ligera y el otro familiar es asintomático (II-5).
En la familia #2 el estudio bioquímico fue normal, excepto en un caso (II-8), que mostró una relación ankirina/(a + b) ligeramente elevada. Los familiares III-7 y III-9 fueron normales. Cinco familiares fueron clasificados como HS típica (III-5, III-8, II-5, II-8 y I-1). Todos presentaron ictericia neonatal y crisis hemolíticas en el curso de procesos infecciosos. La anemia en los 5 casos fue variable, con cifras entre 7 y 10 g/dL y todos requirieron de transfusiones de sangre en alguna ocasión. Excepto la paciente I-1, que presentaba una marcada esplenomegalia y que había sido transfundida en varias ocasiones, los demás fueron esplenectomizados durante la adolescencia y se encuentran asintomáticos. Se demostró un patrón de herencia autosómico dominante, pero no se pudo determinar la alteración bioquímica. ]]>
De acuerdo con los resultados bioquímicos, la familia #3 presenta una deficiencia combinada de Sp + ankirina. El patrón de herencia es autosómico dominante. Se estudiaron 7 miembros de la familia de los cuales II-3, III-1, III-2, III-4 y IV-1 fueron normales y 3 (IV-2, III-3 y II-2) son portadores de la enfermedad. La paciente III-3 presentó ictericia neonatal y a los 36 días de nacida tuvo un descenso de la Hb (5,4 g/dL), por lo que fue transfundida. Se diagnosticó como portadora de HS a los 6 meses de edad. Tuvo múltiples crisis hemolíticas con requerimientos transfusionales periódicos, por lo que fue esplenectomizada a los 4 años y medio de edad. Se clasificó como HS severa. El caso II-2 se diagnosticó durante este estudio. Se clasificó como HS asintomática. Aunque el familiar I-1 no fue estudiado por nosotros, se pudo conocer que es muy sintomática y presenta frecuentemente disminución de las cifras de Hb. El propósito (IV-2) presenta una HS típica.Los resultados del estudio bioquímico de la familia #4 fueron normales. Se observó un patrón de herencia autosómico dominante, pero no se pudo identificar el defecto bioquímico. Dos fueron clasificados como asintomáticos y diagnosticados durante este estudio (III-3, II-5), mientras que el propósito (IV-1) presenta una HS ligera. El caso II-5 presentó hace 20 años una crisis hemolítica en el transcurso de una "hepatitis", la cual fue asociada también con el contacto con un agente químico tóxico y además fue operado hace 5 años de litiasis vesicular. Se clasificó como HS asintomático. La paciente III-3 fue también diagnosticada por nosotros. No tiene antecedentes de ictericia y solo tuvo anemia ligera durante los embarazos. También se clasificó como HS asintomática.
En la familia #5 se estudiaron 11 familiares y en ninguno (II-2, II-3, II-17, II-18, III-2, III-4, III-6, III-7, III-8, III-10, IV-6) se demostró la presencia de la enfermedad, por lo que no pudo precisarse el patrón de herencia. El propósito (IV-5) es muy severo desde el punto de vista hematológico y clínico. Tuvo varios episodios hemolíticos con ictericia, hiperbilirrubinemia con predominio de indirecta y requerimientos transfusionales. De acuerdo con estos datos se clasificó como HS severa. El estudio bioquímico permitió sugerir que el propósito tiene una deficiencia de Sp.
Los datos hematológicos y clínicos del propósito IV-1 (familia #6), permitieron clasificarlo como HS típica. Mantiene anemia, reticulocitosis, bilirrubina aumentada a expensas de la indirecta y esplenomegalia, pero no ha tenido requerimientos transfusionales. Se estudiaron 6 familiares (II-10, II-11, III-2, III-3, III-4, IV-2) y solo uno (II-11) fue diagnosticado con HS. Este paciente no tiene antecedentes de anemia, ictericia ni visceromegalia y fue clasificado como asintomático. Los resultados de los padres del propósito fueron normales, por lo que no se pudo precisar el patrón de herencia. El estudio bioquímico permitió demostrar que varios miembros de la familia tienen deficiencia de banda 3 (IV-1, IV-2, III-2, III-3, III-4), sin embargo, solo el propósito tuvo manifestaciones clínicas.
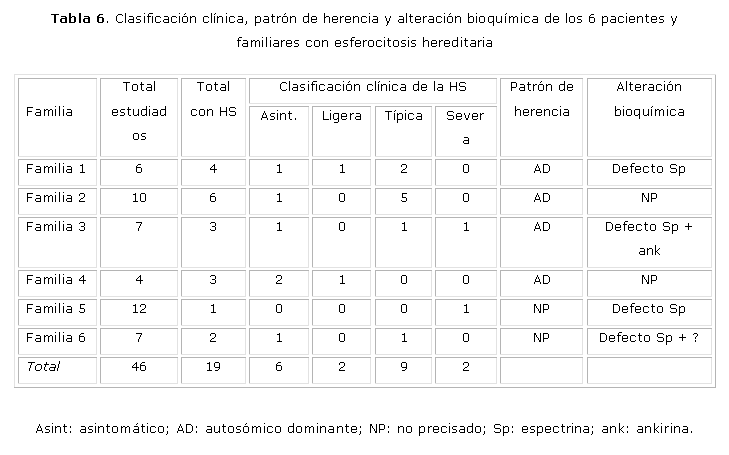
En la tabla 6 se presentan los resultados del estudio desde el punto de vista genético y bioquímico y la clasificación clínica de los casos. Se estudiaron 46 individuos (pacientes y familiares) y de ellos, 19 eran portadores de HS (41,3 %). De acuerdo con la clasificación clínica, 6 (31,6 %) son asintomáticos, 2 (10,5 %) tienen HS ligera, 9 HS típica (47,4 %) y 2 (10,5 %) HS severa. En 4 familias (66,7 %) el patrón de herencia fue autosómico dominante y en 2 no se pudo precisar. En 4 se encontró una defecto de Sp, en una de ellas combinado con defecto de ankirina y en otra asociado posiblemente con una mutación de banda 3 o con un alelo esferocitogénico.
]]>
DISCUSIÓN
Los valores normales de la relación entre las proteínas de la membrana eritrocitaria (Sp a + b, ankirina, banda 3, proteína 4.2) han sido estudiados por diferentes investigadores, y en algunas ocasiones muestran notables diferencias. Esto puede ser debido, fundamentalmente, al empleo de distintas técnicas para la extracción de la membrana eritrocitaria y la electroforesis de las proteínas de membrana, así como a los métodos y programas empleados para la realización de la densitometría de los geles de poliacrilamida. Por ello es siempre conveniente establecer los valores normales en las condiciones de trabajo de cada laboratorio y comparar con los resultados obtenidos por otros investigadores con el empleo de iguales condiciones técnicas. Nuestros valores normales son similares a los señalados por Saad y Miraglia del Giudice.6,20
Familia #1: El cuadro hematológico y clínico de esta familia fue variable; los 4 familiares fueron portadores de HS y todos tenían las cifras de Hb dentro de límites normales al momento del estudio. Sin embargo, 2 presentaban una SH típica, uno HS ligera y el otro HS asintomática. Los 2 casos con HS típica habían sido esplenectomizados. Los resultados bioquímicos permitieron demostrar una deficiencia de Sp. Algunos investigadores han señalado que dentro de una familia el cuadro clínico es similar en todos los miembros afectados y que la severidad de la hemólisis y la respuesta a la esplenectomía es inversamente proporcional a los niveles de espectrina.13,21,22 En esta familia, el cuadro clínico es variable y no se observó una relación entre los niveles de Sp y la severidad de la enfermedad.
Familia #2: La paciente GCPM, aunque siempre ha mantenido cifras de Hb normales, ha presentado en ocasiones reticulocitosis ligera. De los 10 familiares estudiados, 6 son portadores de HS y 5 de ellos fueron clasificados con la forma clínica de HS típica. Todos refieren haber tenido un cuadro similar y fueron esplenectomizados en edades tempranas. Actualmente se encuentran asintomáticos. El estudio de las proteínas de membrana fue normal en todos los casos, por lo que no se pudo precisar la alteración bioquímica responsable de la enfermedad. ]]>
Familia #3: El propósito fue clasificado como HS típica y de los 8 familiares estudiados, 3 son portadores de HS, pero la expresión clínica de cada uno de ellos fue diferente, tal y como encontramos en la familia #1 (un asintomático, uno con HS severa y otro con HS típica). Los casos estudiados tenían una deficiencia de Sp. No se encontró correlación entre la disminución de la Sp y la severidad del cuadro clínico. Sin embargo, el defecto de ankirina (relación ankirina/a + b) se observó solo en los 2 casos que tuvieron manifestaciones clínicas. Estos hallazgos corroboran algunos resultados señalados en la literatura.23,24 Diversos investigadores han planteado que la deficiencia combinada de Sp y ankinira no es fácil de determinar, ya que puede ser enmascarada por la reticulocitosis. Otros señalan que este defecto es más fácil de demostrar en individuos que han sido esplenectomizados.21-23 También se han señalado problemas de sensibilidad de las técnicas empleadas habitualmente. Se ha observado que en esta deficiencia combinada está presente una marcada microcitosis y acantocitosis.21,24-26 En la fijación con glutaraldehído de la paciente III-3 y en el propósito, se observan abundantes microcitos y acantocitos, no así en el caso II-2, lo que se corresponde con lo planteado anteriormente. Es muy probable que la deficiencia de ankirina sea más ligera en el paciente II-2 (el cual presenta también una forma mas benigna de la enfermedad), por lo que es necesario emplear técnicas más sensibles para su detección.
Familia #4: De los 4 miembros de la familia estudiados, 2 tenían la enfermedad y se clasificaron como asintomáticos. Ambos fueron diagnosticados durante este estudio. En esta familia también se observa una diferencia en la expresión del cuadro clínico de la enfermedad. El estudio de las proteínas de la membrana fue normal, por lo que no se pudo precisar el defecto bioquímico.
Familia #5: En esta familia no se pudo precisar el patrón de herencia a pesar de que se estudiaron a 11 individuos. En la literatura se señala que en el 75 % de los casos el patrón de herencia es autosómico dominante, mientras que el resto exhibe una forma autosómica recesiva o la presencia de nuevas mutaciones en los genes de Sp y/o ankirina.22,26 En nuestro caso, que es muy sintomático (HS severa), se puede plantear la presencia de una nueva mutación o de una forma recesiva (heterocigótico de doble componente) con penetrancia incompleta en que ambos padres son heterocigóticos pero no expresan la enfermedad.21,24,27 Este paciente presentó un defecto de Sp y de acuerdo con la literatura, en la forma recesiva la única alteración que se ha encontrado es esta.21,27,28.
Familia #6: En esta familia, en la cual se estudiaron a 6 miembros, solo el abuelo materno (II-11) fue diagnosticado como portador de HS. El resto de los familiares (II-10, III-2, III-3, III-4, IV-2) mostraron un estudio normal. Por tal motivo no fue posible determinar el patrón de herencia. Una hipótesis que explique estos resultados sería que la abuela materna tenga un alelo LE que disminuya la expresión de la enfermedad. De esta forma, la madre hereda la HS del abuelo y el LE de la abuela. Sin embargo, en el propósito podría expresarse la enfermedad si el padre presenta algún defecto de las proteínas de la membrana. En el análisis bioquímico de las proteínas de la membrana eritrocitaria se encontró una deficiencia de banda 3 en varios miembros de la familia. Se podría plantear que el abuelo materno tiene una mutación silente de banda 3 con un alelo LE, asociado con un defecto combinado de Sp y ankirina. La madre sería heterocigótica para esa forma recesiva y además expresa la mutación de la banda 3, pero no tiene manifestación de la enfermedad. El propósito heredaría estos 2 defectos de la madre y una mutación silente esferocitogénica del padre, que conllevaría a un agravamiento del cuadro clínico en comparación con el que expresa el abuelo.19,25,29 Para demostrar esto sería necesario realizar estudios moleculares al paciente y sus familiares.
El porcentaje de las diferentes formas clínicas de la HS encontrado (asintomático 6: 31,6 %; ligera 2: 10,5 %; típica 9: 47,4 %; y severa 2: 10.5 %), es similar a lo señalado en la literatura.2,3,12,21,23
Se observó una gran heterogeneidad en la expresión de la enfermedad en nuestros pacientes desde el punto de vista hematológico y clínico, no solo entre las familias, sino dentro de cada una de ellas. Aunque existen contradicciones en la literatura en este sentido, hay investigadores que plantean que esta variabilidad puede ser debida a una penetrancia incompleta o a la expresión concomitante de un gen modificador (genes esferocitogénicos), lo cual ya ha sido encontrado en algunas familias con deficiencia aislada de Sp y banda 3.29,30 La presencia de otro defecto intrínseco del glóbulo rojo podría incrementar o reducir el fenotipo de la HS y explicar igualmente la heterogeneidad dentro de una misma familia. Este es el caso de la asociación de esta enfermedad con el rasgo b talasémico o la deficiencia de G6PD.31,32 En nuestras familias se descartó esta posibilidad, por lo que pensamos en la posible presencia de otros genes esferocitogénicos responsables de modular la expresión de la HS. ]]>
El estudio de un número mayor de familias así como la introducción de las técnicas de biología molecular, permitirán precisar con exactitud las alteraciones más frecuentes en las proteínas de la membrana eritrocitaria en los pacientes con HS en nuestra población.
REFERENCIAS BIBLIOGRÁFICAS
1. Eber S, Lux SE. Hereditary spherocytosis defects in proteins that connect the membrane skeleton to the lipid bilayer. Sem Hematol 2004;41:118-41.
]]>
2. Gallagher PG, Jarolim S. Red cell membrane disorders. En: Hematology, Basis Principles and Practice. 4 ed Philadelphia: WB Saunders; 2005. p. 669-91.
3. Gallagher PG. Update on the clinical spectrum and genetics of red blood cell membrane disorders. Curr Hematol Rep 2004;3:85-91.
4.------. Red cell membrane disorders. Hematology Am Soc Hematol Educ Program 2005:13-18.
5. Gallagher PG, Forget BG. Hematologically important mutations: Spectrin and ankirin variants in hereditary spherocytosis. Blood Cells, Molecules and Diseases 1998;24:539-43.
6. Saad ST, Costa FF, Vicentim DL, Salles TS, Pranke PH. Red cell membrane protein abnormalities in hereditary spherocytosis in Brazil. Br J Haematol 1994;88:295-9.
]]>
7. Bolton-Maggs PHB. Hereditary spherocytosis; new guidelines. Arch Dis Child 2004;89:809-12.
8. An X, Mohandas N. Disorders of red cell membrane. Brit J Haematol 2008;141:367-75.
9. Tamary H, Aviner S, Freud E, Miskin H, Krasnov T, Schwarz M, et al. High incidence of early cholelithiasis detected by ultrasonography in children and young adults with hereditary spherocytosis. J Pediatr Hematol Oncol 2003;25:952-4.
10. Del Giudice EM, Perrotta S, Nobili B, Specchia C, d´Urso G, Iolascon A. Coinheritance of Gilbert syndrome increases the risk for developing gallstones in patients with hereditary spherocytosis. Blood 1999;94:2259-62.
11. Fairbanks G, Steck TL, Wallasch DFH. Electrophoretic analysis of the major polypeptides of the human erythrocyte membrane. Biochemistry 1971;10:2606-17.
]]>
12. Hassoun H, Palek J. Hereditary spherocytosis: A review of the clinical and molecular aspects of the disease. Blood Rev 1996;10:129-47.
13. Tracy ET, Rice HE. Partial splenectomy for hereditary spherocytosis. Pediatr Clin N Am 2008;55:503-19.
14. Lewis SM, Bain BJ, Bates I. Dacie and Lewis Practical Haematology. 9 ed. London: Churchill Livingstone; 2001.
15. Saenz GF, Moreira J. Laboratorio Hemoglobinopatías. San José, Costa Rica: Manual Latinoamericano; 1980.
16. Zanella A, Izzo C, Rebulla F, Zanuso F, Perroni L, Sirchia G. Acidified glycerol lysis test: A screening test for spherocytosis. Brit J Haematol 1980;45:481-5.
]]>
17. Dhermy D, Garbaz M, Lecomte MCh, Feo C, Bounier O, Chaveroche I, et al. Hereditary ellyptocytosis: Clinical, morphological and biochemical studies of 38 cases. Nouv Rev Fr Hematol 1986;26:129-40.
18. Sonnenwirth AC, Jarrett L. Métodos y diagnósticos de laboratorio clínico. La Habana: Ed Científico-Técnica; 1993.
19. Lecomte MCh, Garbaz M, Grandchamp C, Feo C, Gautero H, Devaux O, et al. SpaI/78. A mutation of the aI spectrin domain in a white kindred with HE and HPP phenotypes. Blood 1989;74:1126-33.
20. Del Giudice ME, Perrota S, Pinto S, Cappellini MD, Fiorelli G, Cutillo S, et al. Hereditary spherocytosis characterized by increased spectrin/band 3 ratio. Brit J Haematol 1992;80:133-6.
21. Iolascon A, del Giudice ME, Perrota S, Allosio N, Morle L, Delaunay J. Hereditary spherocytosis: From clinical to molecular defects. Haematologica 1998;83:240-57.
]]>
22. Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet 2008;372:1411-26.
23. Delaunay J. Genetic disorders in the red cell membrane. Clin Rev Oncol Hematol 1995;19:79-110.
24. Saada V, Cynober T, Brossard Y, Schischmanoff PO, Sender A, Cohen H, et al. Incidence of hereditary spherocytosis in a population of jaundiced neonates. Pediatr Hematol Oncol 2006;23:387-97.
25. Hassoun H, Vassiliades JN, Murray J, Njolstad PR, Rogus JJ, Ballaas SK, et al. Characterization of the underlying molecular defect in hereditary spherocytosis associated with spectrin deficiency. Blood 1997;90:398-406.
26. Del Giudice ME, Francese M, Nobili B, Morlé L, Cutillo S, Delaunay J, et al. High frequency of the novo mutations in ankirin gene (ANK1) in children with hereditary spherocytosis. J Pediatr 1998;132:117-20.
]]>
27. Agre P, Orringer EP, Bennett V. Deficient red-cell spectrin in severe, recessively inherited spherocytosis. N Engl J Med 1982;306:1155-9.
28. Herrera M, Estrada M. Esferocitosis hereditaria: aspectos clínicos, bioquímicos y moleculares. Rev Cub Hematol Inmunol Hemoter 2002; Disponible en: <http://scielo.sld.cu/scielo.php?script=sci_arttex /&pid=S0864-ISSN 0864-0289.
29. Alloisio N, Maillet P, Carré G. Hereditary spherocytosis with band 3 deficiency. Association wit a nonsense mutation of the band 3 gene (Allele Lyon) and aggravation by low-expression allele occurring in trans (Alelle Genas). Blood 1996;88:1062-9.
30. Wichterle H, Haanspal M, Palek J, Jarolim P. Combination of two mutant a spectrin alleles underlies a severe spherocytic hemolytic anemia. J Clin Invest 1996;98:2300-7.
31. Del Giudice ME, Perrota S, Nobili B, Pinto L, Cutillo L, Iolascon A. Coexistence of hereditary spherocytosis (HS) due to band 3 deficiency and b thalassaemia trait: Partial correction for HS phenotype. Br J Haematol 1993;85:553-7.
]]>
32. Affinito F, Calabro V, Cappellini MD. G6PD deficiency and red cell membrane defects: Aditive or synergistic interaction in producing chronic haemolytic anaemia. Br J Haematol 1994;87:146-52.
Recibido: 14 de octubre del 2009.
Aprobado: 26 de octubre del 2009.
]]>
DraC. Marianela Estrada del Cueto. Instituto de Hematología e Inmunología. Apartado 8070. Ciudad de La Habana, CP 10800, Cuba. Tel (537) 643 8268, 643 8695, Fax (537) 644 2334. e-mail: ihidir@hemato.sld.cu , mestrada@hemato.sld.cu ]]>