
ARTÍCULO DE REVISIÓN
Hemofilia: aspectos históricos y genéticos
]]>
Hemophilia: historical and genetic aspects
Dra. Dunia Castillo-González
Instituto de Hematología e Inmunología. La Habana, Cuba. ]]>
]]>
]]>
RESUMEN
La hemofilia es un trastorno hemorrágico con disminución o ausencia de la actividad procoagulante del factor VIII o del IX. Las primeras descripciones de esta enfermedad son tan antiguas como la propia humanidad. A lo largo de los años, la hemofilia ha sido nombrada "enfermedad real" debido a que la padecieron diversos miembros de las familias reales europeas. En la actualidad, mediante estudios moleculares, se encontró el defecto genético causante de la enfermedad en los varones hemofílicos de la familia de la Reina Victoria y se encontró que sus descendientes padecieron una hemofilia B severa. El fenotipo de esta enfermedad es hemorrágico; se observan sangramientos en diversos sitios de la economía condicionados fundamentalmente por los niveles del factor deficiente. Existen otros factores que intervienen en las características fenotípicas variables de estos pacientes, entre ellos: las características intrínsecas de los factores VIII/IX, la presencia de genes modificadores y factores ambientales que influyen sobre la severidad de la enfermedad. Se revisa la correlación genotipo-fenotipo en esta enfermedad mediante las mutaciones más frecuentes en cada tipo de hemofilia. En cuanto a la presencia de los inhibidores, se destacan las evidencias actuales en relación con los factores de riesgo relacionados con su aparición y los aspectos moleculares presentes en la variante de hemofilia B Leyden.
Palabras clave: hemofilia, genética, mutaciones.
]]>
ABSTRACT
Hemophilia is a hemorrhagic disorder characterized by decreasing or absence of the procoagulant activity in factor VIII or IX. First descriptions of this disease are as old as mankind itself. During time, hemophilia has been called "royal disease" since different members of European royal families suffered from it. Currently, by molecular studies, it was found the causing genetic defect of this disease in hemophilic male members of Queen Victoria´s family; and it was found that her descendants suffered a severe hemophilia B. This disease phenotype is hemorrhagic; bleeding in different sites is observed which are mainly conditioned by the levels of the deficient factor. There are other factors participating in the variable phenotype characteristic of these patients, such as: intrinsic characteristics of factors VIII/IX, the presence of modifying genes and environment factors which influence on this disease severity. It is revised here the correlation genotype-phenotype in this disease through the most frequent mutations in each type of hemophilia. Concerning the presence of inhibitors, it is highlighted the current evidences in relation with risk factors related to its emergence and molecular aspects present in hemophilia variant B Leyden.
Key words: hemophilia, genetic, mutations.
]]>
INTRODUCCIÓN
La hemofilia es un trastorno hemorrágico con disminución o ausencia de la actividad procoagulante del factor VIII o del IX. La incidencia es casi constante en las diferentes poblaciones, para la hemofilia A (HA) 1/5-10 000 varones y la hemofilia B (HB) 1/60 000 varones. Es la segunda enfermedad genética hemorrágica más frecuente, después de la enfermedad de von Willebrand (EvW), y la más frecuente de las entidades hereditarias ligadas al cromosoma X.1
ANTECEDENTES HISTÓRICOS ]]>
Las primeras descripciones de esta enfermedad son tan antiguas como la propia humanidad. Los primeros indicios se remontan a los papiros egipcios y en el libro antiguo sagrado de los judíos, el Talmud, en el siglo II antes de Jesucristo. En este libro se describía cómo algunos varones luego de ser circuncisos, presentaban hemorragias agudas que los llevaban a la muerte. Los rabinos no sabían a qué se debía esa anomalía, pero fueron conscientes de que estos problemas del sangrado solo ocurrían en ciertas familias. Por esta razón, el patriarca Rabbi Judah estableció que los terceros varones pertenecientes a una familia en la que los 2 hijos anteriores hubiesen muerto desangrados, quedaban exentos de este proceder. Posteriormente, el rabino Simón Ben Gamaliel impidió que un niño fuese circunciso porque los hijos de las 3 hermanas de la madre se habían desangrado hasta morir.2
El médico árabe Albucasis, en el siglo XI, también describió una familia en la que los varones murieron luego de una lesión trivial. El médico hebreo Moisés Maimónides, en el siglo XII, descubrió que si los niños tenían hemofilia eran las madres las que la transmitían, por lo que aplicó una ley nueva "si una madre tenía hijos con este problema de sangrado y ella se volvía a casar, ninguno de sus nuevos descendientes varones deberían ser circuncisos", lo que demostró ser un reconocimiento temprano de la naturaleza hereditaria del trastorno.2
Las primeras referencias en Europa fueron en 1525, en Italia, por Alejandro Benedicto. En el año 1796 apareció comunicado en la Gaceta de Salem, Massachusetts, el primer caso en Norteamérica que no constituyó una referencia científica, pero se reflejaba el caso de un joven de 19 años que sangró profusamente.3
Años más tarde, en 1803, apareció la primera descripción científica moderna de la hemofilia en Norteamérica en el tratado titulado "Recuento de una disposición hemorrágica existente en ciertas familias", en el que se describieron 3 aspectos básicos de la enfermedad, la tendencia hereditaria a las hemorragias en varones y se rastreó la genealogía de la familia hasta alrededor de 1720.3,4
En 1813, en la revista New England Journal of Medicine Surgery, se publicó el primer árbol genealógico en hemofilia "An account of a remarkable haemorrhagic disposition existing in many individuals of the same family".3,4
No obstante, el término hemofilia, del griego hemo -sangre- y filia -amor- apareció por primera vez en una descripción escrita en 1828 titulada "Über die haemophilie oder die erbliche Anlage zu todlichen Blutungen".3
A lo largo de los años, la hemofilia ha sido nombrada enfermedad real debido a que la padecieron diversos miembros de la nobleza europea. La Reina Victoria no tenía antepasados con este trastorno, pero poco después del nacimiento de su último hijo, Leopoldo, en 1853, se evidenció que padecía hemofilia, por lo que constituyó un ejemplo de que la hemofilia podía aparecer por una nueva mutación, o sea, sin hallazgos previos en familiares. Leopoldo murió a los 31 años a causa de una hemorragia intracerebral después de una caída.
Dos de las hijas de la Reina Victoria, Alice y Beatrice, fueron portadoras de la hemofilia; ellas transmitieron el padecimiento a diversas familias de la realeza de Europa, incluida España y Rusia. Alexis Nikolayevich Romanov, nacido en 1904, hijo del zar Nicolás II de Rusia y Alexandra de Hasse, nieta de la Reina Victoria de Inglaterra, ha sido la persona más famosa afectada por esta enfermedad.2,4 En la actualidad, ninguno de los descendientes de la gran familia real europea padece la enfermedad. Recientemente, a partir del descubrimiento de las tumbas del zar Nicolás II en 1991, de la princesa Anastasia y el zarevich Alexis, en el 2007, se pudo estudiar el ADN de ambos hermanos.5 Se encontró que el pequeño infante presentaba una HB severa y su hermana era portadora de la enfermedad. Por estudios moleculares se encontró el defecto genético causante de la enfermedad, una mutación intrónica A>G en un sitio de empalme en una posición localizada 3 pares de bases antes del exón 4. Esta sustitución crea una nueva zona crítica de empalme que produce una lectura aberrante. Se forma un codón de terminación temprana y la síntesis de una proteína muy anómala. Estos resultados evidenciaron que los varones hemofílicos de la familia de la Reina Victoria y sus descendientes padecieron una HB severa. Esta mutación ha sido descrita en otros individuos no relacionados con esta familia y también presentaban la enfermedad en forma severa.5
Como se ha demostrado, en el siglo XIX el patrón de herencia característico se fue esclareciendo a partir de las descripciones de la enfermedad. En 1947, el argentino Alfredo Pavlovsky mediante sus estudios, sugirió que la hemofilia no era una enfermedad homogénea.6 Cinco años más tarde, Rosemary Biggs y otros, describieron la enfermedad de Christmas o hemofilia B.7 ]]>
CARACTERIZACIÓN FENOTÍPICA DE LA ENFERMEDAD
El fenotipo de esta enfermedad es hemorrágico, se observan sangramientos en diversos sitios de la economía condicionados fundamentalmente por los niveles del factor deficiente. Este trastorno afecta casi exclusivamente a varones y las portadoras son las mujeres, salvo algunas excepciones en que pueden aparecer mujeres hemofílicas debido a fenómenos como la inactivación desfavorable del cromosoma X, isodisomía o la concomitancia con un síndrome Turner, entre otras situaciones clínico-genéticas.2
Las hemorragias músculo-articulares constituyen el sello distintivo de esta entidad, donde las grandes articulaciones suelen ser diana de sangramientos frecuentes; las más afectadas son: rodilla, codo, tobillo, hombro, cadera, e importantes grupos musculares como: muslo (cuadríceps femoral), pantorrilla (gastrocnemios y sóleos), antebrazo, brazo (bíceps y tríceps), glúteos mayores y de singular importancia, la hemorragia del músculo psoas ilíaco por la forma de presentación y la gravedad que conlleva un sangramiento en esta estructura retroperitoneal.2 Otras manifestaciones clínicas son las hemorragias observadas en diferentes regiones del cuerpo humano, las del sistema nervioso central, que pueden ser intraparenquimatosas, subaracnoideas o intramedulares, que producen una mortalidad elevada si no son tratadas oportunamente;8 las pulmonares del parénquima, pleuras o en forma de hemoptisis;9 las mediastinales,10 urinarias, digestivas, oftálmicas, de la mucosa oral, entre otras menos frecuentes.2
]]>
ASPECTOS GENÉTICOS
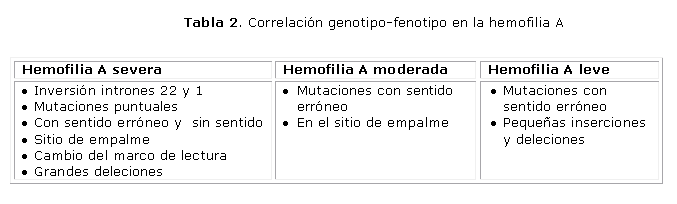
Esta enfermedad está caracterizada por una heterogeneidad fenotípica y alélica. Más de 1 000 mutaciones han sido descritas para cada hemofilia y siempre causan un fenotipo hemorrágico.11,12 En las tablas 1 y 2 se muestran las mutaciones y su relación con la severidad del fenotipo, tanto para los pacientes con HA o HB.
Mutaciones puntuales
En el registro más reciente de mutaciones en la HB se encuentran 1 918 pacientes. Se incluyen deleciones de tamaño variable y cerca de 1 500 mutaciones puntuales: con sentido erróneo y sin sentido, en el sitio de empalme, en el promotor, entre otras localizaciones.11 En el caso de la HA, las mutaciones más frecuentemente observadas son las inversiones, las grandes deleciones y las mutaciones puntuales (con sentido erróneo y sin sentido).12 ]]>
La gravedad de las manifestaciones clínicas va a estar dada fundamentalmente por los niveles plasmáticos del factor VIII (FVIII) y IX (FIX), que han resultado de la mutación que se presenta. Según recomendaciones del Subcomité de los Factores VIII/IX del Comité Científico de la Sociedad Internacional de Hemostasia y Trombosis,13 la hemofilia se clasifica en relación con los niveles de actividad de los factores de la coagulación en: severa (actividad menor del 1 %), moderada (actividad entre el 1-5 %) y leve (actividad mayor del 5 y hasta el 40 %). El nivel del factor deficitario es similar en todos los miembros afectados de una familia.Está demostrado que para definir un estado de gravedad no solo es el nivel de factor el que lo condiciona, sino otros factores que determinarán la capacidad de cada paciente para generar trombina.2
Se han estudiado varios factores que podrían influir en las características fenotípicas variables de estos pacientes; entre ellas se encuentran: las características intrínsecas de los factores VIII/IX, la presencia de genes modificadores de los genes de estos factores con efectos epistáticos que influyen sobre la severidad de la enfermedad, por ejemplo, los genes relacionados con ciertos estados trombofílicos.14 En relación con este último aspecto, se ha comunicado que niños hemofílicos con la presencia de mutaciones en genes protrombóticos, han tenido una mejor evolución clínica en cuanto a menos eventos hemorrágicos e inicio más tardío de ellos. Otros factores relacionados con el aclaramiento más rápido de los factores de la coagulación, como sucede en los niños, han sido la farmacocinética del FVIII, pacientes con grupo sanguíneo 0 o con niveles bajos del factor von Willebrand (FvW).15
Algunos grupos de trabajo han investigado la influencia de los factores relacionados con la fibrinólisis. Se ha descrito que pacientes con niveles aumentados del activador tisular del plasminógeno presentarían una fibrinólisis aumentada con incremento de la severidad hemorrágica.2
La heterogeneidad del fenotipo, además, se manifiesta de otras disímiles formas; el momento de inicio de las hemorragias suele variar aún en los pacientes con igual severidad. Hay pacientes que han presentado eventos hemorrágicos desde la etapa neonatal, durante el momento del parto, otros en la etapa de lactantes cuando son sometidos a las vacunaciones o punturas. En los niños,los sangramientos más observados son los pequeños hematomas subcutáneos y la ruptura del frenillo labial, que ocurren cuando comienzan a deambular. Otro aspecto a considerar es la variación en la frecuencia de los sangramientos y la afectación que se produce en las articulaciones con el consecuente daño articular.15
Para individualizar mejor el tratamiento los pacientes con hemofilia, se han intentado clasificar considerando diversos criterios. Se han denominado pacientes intensamente y no intensamente hemorrágicos según las hemartrosis que presenten por año, la cantidad de tratamiento sustitutivo necesario para tratar los eventos hemorrágicos, la respuesta al tratamiento y el inicio de los eventos hemorrágicos.
Otros factores descritos han sido los marcadores de angiogénesis y de respuesta inflamatoria.14,15 Se ha sugerido, a partir de investigaciones en la artritis reumatoidea, que los niveles altos de interleucina (IL) 10 e interferón-g pudieran tener un efecto protector contra el deterioro articular. Otros estudios evidencian que el aumento de las citoquinas proinflamatorias IL1b, IL6, factor de necrosis tumoral a (FNTa) en la membrana sinovial de las articulaciones de los hemofílicos unido con el hierro, monocitos y fibroblastos locales, son capaces de generar superóxidos y radicales libres que pueden inducir la expresión de diversos oncogenes, entre ellos el c-myc, lo que ejerce un efecto sinérgico en la proliferación y daño articular observado en este grupo de pacientes.15 Los marcadores de angiogénesis han sido igualmente estudiados, entre ellos, el factor de crecimiento del endotelio vascular, que se ha observado aumentado en pacientes con artropatía crónica.15
Una complicación seria y no poco frecuente que caracteriza el fenotipo de estos pacientes, lo constituyen los inhibidores (anticuerpos contra los factores VIII/IX infundidos), que son inducidos por la terapia sustitutiva con una incidencia variable según diferentes estudios. Estos aloanticuerpos inhibidores tipo IgG fundamentalmente clase 4, son anticuerpos neutralizantes que interfieren en la función de los FVIII y FIX con implicaciones clínico-terapéuticas importantes como la dificultad para el remplazo de forma terapéutica de los factores de la coagulación. Está demostrado que los epítopes antigénicos son específicos y están localizados fundamentalmente en las dominios A2, C2, y C1 del FVIII.16-19 (Fig.).
Esta complicación se observa con mayor frecuencia en la HA, entre el 12-35 %, y en la HB es significativamente más baja, entre el 1-4 %.2,16,17 La presencia de estos inhibidores agrava la evolución clínica de los pacientes, aumenta los costos del manejo de los eventos hemorrágicos y eleva ampliamente la morbimortalidad. En los pacientes severos se observa con mayor incidencia, alrededor del 50 %, y en los leves y moderados disminuye de forma considerable la incidencia de aparición a valores entre el 3-13 %.17 A principios de la década de los 90 en Cuba, se realizó un estudio sobre la evolución de los inhibidores en los pacientes con hemofilia A y B; se detectó la presencia de estos anticuerpos en el 13,6 % de los pacientes con HA y el 1,4 % de los pacientes con HB.18 ]]>
Existen diferentes factores de riesgo que parecen relacionarse con la formación de inhibidores que pueden ser genéticos y no genéticos. Dentro de los primeros encontramos el tipo de defecto molecular, etnicidad, inmunofenotipo HLA, defecto molecular de genes de citoquinas e historia familiar. En los de tipo no genéticos se encuentran: el tipo de producto utilizado, la intensidad de las primeras infusiones, la edad de la primera infusión, el número y patrón de exposiciones al factor exógeno, el método de infusión, el producto utilizado, la transfusión sanguínea previa al remplazo del factor, las enfermedades febriles concomitantes y las vacunaciones.15-20Desde el punto de vista de mutaciones asociadas con estos inhibidores, se observan con mayor frecuencia la inversión del intrón 22, aproximadamente en el 50 % de los casos; menos frecuentes están la inversión del intrón 1, las grandes deleciones y las mutaciones sin sentido,16,17 como describimos anteriormente. La historia familiar se ha demostrado que triplica la probabilidad de que aparezcan los inhibidores. En una pequeña investigación controlada realizada en Estados Unidos, donde se estudiaron 78 pacientes afronorteamericanos con igual severidad y patrón de mutaciones, se demostró mayor prevalencia de 2 haplotipos relacionados con el FVIII.21 Se evidenció que en este grupo de pacientes, los haplotipos predominantes eran diferentes a los encontrados en los enfermos de origen caucásico. Los productos de remplazo utilizados fueron igualmente estudiados y se observó que en ellos predominaban los haplotipos descritos en los pacientes de origen caucásico.17,21 Aunque este fue un estudio pequeño, trató de explicar las diferencias encontradas en pacientes de orígenes africanos y caucásicos.
Dentro del grupo de los factores no genéticos, en la actualidad resultan de gran interés los estudios que analizan el tipo de producto sustitutivo utilizado y el comienzo precoz de la profilaxis primaria. En relación con la terapia de remplazo, se ha demostrado que aquellos pacientes que utilizan los productos de origen plasmático, que poseen cantidades suficientes de FvW, tienen menos probabilidad de presentar inhibidores. Se ha encontrado que esta proteína, esencial para la coagulación sanguínea por sus aportes en la adhesión plaquetaria, como transportador y protector del FVIII de la proteólisis enzimática, desempeña un papel preponderante.22 Estas investigaciones han demostrado que el FvW tiene una asociación directa con la generación de trombina. Aquellos concentrados de FVIII de pureza intermedia, como el Humate P, son capaces de generar mayor cantidad de trombina que aquellos productos que posean trazas de esta glicoproteína.22
Se han postulado 2 mecanismos para explicar este fenómeno, el enmascaramiento de epítopes antigénicas y la protección del FVIII de las células dendríticas. El FVIII presente en los concentrados de factores VIII/FvW es menos susceptible de ser neutralizado por anticuerpos dirigidos contra la cadena ligera del FVIII y el dominio C2, sitios blanco de acción de los inhibidores. In vitro se ha demostrado que el FvW actúa como una chaperona inmunoprotectora del factor VIII al impedir la activación de efectores inmunes.22 Experimentos in vitro, han demostrado que el FVIII o péptidos de este actúan como antígeno y son reconocidos por las células presentadoras de antígenos, las células dendríticas; estas presentan los antígenos a las células T CD4+ auxiliadoras con la consiguiente activación de la respuesta inmune mediada por anticuerpos. Se ha demostrado, además, que cuando se han utilizado ligandos que bloquean estos linfocitos T, se ha abolido la respuesta inmune en este grupo de pacientes.16,22,23 Desafortunadamente, estas observaciones no se han podido demostrar aún in vivo.
En el caso de la HB, donde la incidencia de inhibidores es muy baja, el mecanismo involucrado no es similar al descrito en la HA. El FIX es una proteína pequeña capaz de presentarse en el espacio extravascular y activar a los mastocitos y los mecanismos de hipersensibilidad mediado por la IgE. Normalmente, los pacientes con HB se han expuesto a productos menos puros que los hemofílicos A y las reacciones alérgicas son las más frecuentemente asociadas. En relación con las mutaciones, se observan asociadas sobre todo con las grandes deleciones.24
Otros factores que influyen en la evolución de los pacientes son aquellos denominados "ambientales", que están relacionados con el estilo de vida y hábito corporal. Aquellos pacientes con un comportamiento más dinámico desde el punto de vista físico, suelen tener eventos hemorrágicos más frecuentes, pero cuando esa actividad física es reiterada, mantenida y bien dirigida, es un factor positivo para que los músculos se fortalezcan y ayuden a disminuir las hemorragias articulares.14 Por nuestra parte, siempre exhortamos a los pacientes a mantener una vida libre de sedentarismo, ya que de esa forma las hemorragias suelen ser leves y se solucionan con más rapidez.
En estudios realizados en gemelos monocigóticos se ha observado discordancia con el tiempo de aparición de los eventos hemorrágicos, farmacocinética de los factores de la coagulación y la respuesta al tratamiento.2,16 Las evidencias actuales demuestran que los factores no genéticos desempeñan un papel muy importante en la aparición de los inhibidores. Diversos grupos de trabajo han tratado de estratificar a los pacientes hemofílicos para encontrar grupos de riesgo y establecer tempranamente un tratamiento diferenciado que disminuya la aparición de esta complicación relacionada con la enfermedad.20,25,26
Desde el punto de vista terapéutico, los pacientes con inhibidores se clasifican en respondedores altos (AR) y bajos (BR) en dependencia de la respuesta de memoria inmunológica que presenten. Los AR son aquellos que responden de forma rápida con títulos altos, mayores de 5 unidades Bethesda (UB/mL) del inhibidor frente al estímulo antigénico y no tienden a desaparecer con el tiempo, mientras que en los BR la respuesta inmunológica suele ser débil, lenta, no siempre constante, que en ocasiones puede desaparecer; desde el punto de vista del laboratorio, y los títulos son inferiores a 5 UB/mL.17,19
]]>
CORRELACIÓN GENOTIPO-FENOTIPO
Esta relación se expresa entre el tipo y posición de la mutación que ocurre y la severidad de la enfermedad.27 Las variaciones de los genes de los FVIII y FIX son pocas entre una persona y otra. El gen del FVIII es uno de los más grandes descritos hasta el momento, si se considera su tamaño, las probabilidades de mutaciones son muy altas y de ahí la heterogeneidad fenotípica tan grande.
Como vimos anteriormente, la probabilidad de desarrollar inhibidores está relacionada, entre otras causas, con el tipo de mutación presente. Por lo general se asocia con aquellas donde no se produce la proteína o está muy truncada, como las grandes deleciones de uno o más exones, mutaciones sin sentido en regiones críticas como dominios A2, C1 y C2 y las inversiones como la del intrón 22. Las mutaciones que menos se asocian con la formación de anticuerpos son las mutaciones puntuales, donde la producción, aunque mínima de los FVIII y FIX, se mantiene.27 Generalmente estos pacientes poseen un fenotipo severo.
Desde hace años se ha reconocido el término de inhibidor transitorio, como aquel que aparece generalmente en bajos títulos, después de un tratamiento intensivo y con tendencia a desaparecer en el tiempo sin un tratamiento específico. Ellos representan el 30-55 % de los inhibidores;17-19 sin embargo, no se han encontrado mutaciones específicas relacionadas con esta presencia. En los pacientes con hemofilia leve y moderada es rara la aparición de los inhibidores. En estos casos se ha visto asociada con mutaciones puntuales con sentido erróneo.27
Un caso distintivo que no se parece al patrón definido previamente en la evolución de la hemofilia es la reconocida como HB Leyden, descrita por Veltkamp y otros en 1970.28 Esta variante, descrita en la ciudad homónima de Holanda, es un caso particular de hemofilia donde el nivel del FIX tiene valores muy bajos o moderadamente bajos en la infancia y se elevan ostensiblemente hacia la pubertad y la adultez.
Este fenotipo se ha precisado en 81 familias no relacionadas; el diagnóstico se confirma con una historia pormenorizada de los síntomas y evolución de la enfermedad y es comprobada por estudio de mutaciones por ensayos de biología molecular. Hasta el 2009 se habían comunicado 14 mutaciones puntuales y deleciones en una pequeña región de 50 pares de bases, entre los nucleótidos -34 al +19 (+19, +13, -5, -6, -20, -26) del promotor del FIX. Esta zona es reconocida como región específica de Leyden (LSR, del inglés Leyden specifical region).28-30 El primer paciente descrito presentó una sustitución puntual de timina por adenina en la posición -20.30
En la actualidad se investiga acerca del origen de la mejoría significativa de los síntomas hemorrágicos en esta rara variante de HB. Algunos estudiosos del tema plantean que el promotor del FIX responde a la testosterona y que puede aumentar las concentraciones del factor durante la pubertad en los pacientes con la mutación Leyden. Ello sugiere que este promotor contiene un elemento de respuesta a andrógenos (ARE) en las posiciones -36 a -22, que sería dependiente de este aumento hormonal durante la pubertad.31,32 Estos hallazgos no se corresponden con la variante HB Brandenburg, en la que la mutación ocurre en el nucleótido -26 (G-26 C) donde se encuentra el ARE y esta variante no tiene aumento de los niveles del FIX durante la pubertad. ]]>
Por estudios recientes se ha observado que el LSR se une con 3 factores de transcripción nuclear: el HNF-4, el UKP-6 y el C/EBP; las mutaciones descritas en la HB Leyden causan interrupción de la unión a HNF-4 y a C/EBP, lo que ocasiona enfermedad severa en la niñez. La pérdida de la unión a UKP-6 causa sintomatología leve. Estudios en ratones transgénicos demostraron que la mejoría en la sintomatología no era específicamente dependiente del aumento de las hormonas masculinas en la pubertad.Existen mecanismos importantes, como el elemento de estabilidad relacionado con la edad (ASE) y el elemento de incremento relacionado con la edad (AIE). El ASE tiene un papel significativo en la regulación específica de tejidos y se une a un factor de transcripción nuclear multifuncional Est-1, ya reconocido con anterioridad por su participación en procesos de carcinogénesis como protoncogen, y que en hallazgos recientes se comprobó su nuevo papel en la regulación fisiológica de la expresión de los genes relacionados con la edad.33 En la pubertad, los mecanismos mediados por ASE/AIE comienzan a remplazar los mecanismos negativos mediados por LSR, el que es esencial en la expresión del gen durante la etapa perinatal y prepuberal.
Existen relaciones recíprocas entre los mecanismos de inicio y apagado de regulaciones en la expresión del gen del factor IX. Estos procesos +ASE/-AIE son complementarios y los incrementos relacionados con la edad en la unión ASE-Est-1 corren paralelos a la regulación positiva de la expresión del gen del FIX.33
La recuperación fisiológica de la expresión del gen del FIX está en relación con la hormona de crecimiento en ambos sexos. Estudios en animales transgénicos hipofisectomizados demostraron que al disminuir severamente los niveles de la hormona de crecimiento, disminuyen los valores del FIX y la rápida inyección intraperitoneal recupera los valores normales en términos de pocos días.33,34 Se han descrito portadoras de las mutaciones de la HB Leyden con niveles bajos en la niñez con manifestaciones hemorrágicas después de eventos quirúrgicos.34 El estudio de estas portadoras permitirá estudiar si los aumentos de los valores del FIX están en relación con los andrógenos solamente o con la hormona del crecimiento, como indican los últimos estudios, o si las hormonas femeninas son también importantes.34
Como se observa, muchos aspectos genéticos se han esclarecido con el desarrollo de los estudios moleculares en los últimos años, pero aún quedan otros por dilucidar que explicarán las diferencias en la expresión fenotípica de los pacientes hemofílicos.
REFERENCIAS BIBLIOGRÁFICAS ]]>
1. Mannucci PM. Ham-Wasserman Lecture: hemophilia and related bleeding disorders: a story of dismay and success. Hematology Am Soc Hematol Educ Program. 2002:1-9. PMID: 12446416.
2. Bolton-Maggs P, Pasi JK. Haemophilias A and B. Lancet. 2003 May 24;361(9371):1801-9.
3. Ingram GI. The history of haemophilia. J Clin Pathol. 1976 Jun;29(6):469-79.
4. Giangrande P. Cronología de la hemofilia [internet]. Montréal, Québec: World Federation of Hemophilia, 2007 [citado 15 sept 2010]. Disponible en: http://www.wfh.org/index.asp?lang=EN
5. Lannoy N, Hermans C. The royal disease-haemophilia A or B? A haematological mystery is finally solved. Haemophilia. 2010 Nov;16(6):843-7. doi: 10.1111/j.1365-2516.2010.02327.
6. Pavlovsky A. Contribution to the pathogenesis of hemophilia. Blood. 1947 Mar;2(2):185-91.
7. Biggs R, Douglas AS, Mac Farlane RG, Dacie JV, Pitney WR, Merskey. Christmas disease: a condition previously mistaken for haemophilia. Br Med J. 1952 Dec 27;2(4799):1378-82.
]]>
8. Balkan C, Kavakli K, Karapinar D. Spinal epidural haematoma in a patient with haemophilia B. Haemophilia. 2006 Jul;12(4):437-40.
9. Girolami A, Vettore S, Scandellari R, Allemand E, Girolami B. About the rarity of haemoptysis in congenital bleeding disorders. A report of five cases. Haemophilia. 2009 May;15(3):825-7.
10. Castillo D, Almagro D, Machín S, Nuñez A, Macías I, Nordet I, et al. Hemorragias en sitios poco usuales en pacientes con hemofilia. XXI Congreso del grupo CLAHT y XX Congreso de la Sociedad Venezolana de Hematología. Isla de Margarita, Venezuela 2-5 Octubre 2009. (P055)
11. Giannelli F, Green PM, Sommer SS, Poon M, Ludwig M, Schwaab R, et al. Haemophilia B: database of point mutations and short additions and deletions—eighth edition. Nucleic Acids Res. 1998 Jan 1;26(1):265-8. ]]>
12. Kemball-Cook G, Tuddenham EG, Wacey AI. The factor VIII structure and mutation resource site: HAMSTeRS version 4. Nucleic Acids Res. 1998 Jan 1;26(1):216-9.
13. White GC II, Rosendaal F, Aledort LM, Lusher JM, Rothschild C, Ingerslev J. On behalf of the Factor VIII and Factor IX Subcommittee. Definitions in hemophilia recommendation of the Scientific Subcommittee on Factor VIII and Factor IX of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 2001 Mar;85(3):560.
14. Van den Berg HM, De Groot PHG, Fischer K. Phenotypic heterogeneity in severe hemophilia. J Thromb Haemost. 2007 Jul;5 (Suppl 1):151-6.
15. Jayandharan GR, Srivastava A. The phenotypic heterogeneity of severe hemophilia. Semin Thromb Hemost. 2008 Feb;34(1):128-41.
16. Goudemand J. Inhibitor developments in haemophilia A: the role of von Willebrand factor/factor VIII concentrates. Haemophilia. 2007 Dec;13 (Suppl 5):47-51.
17. Dimichele D. Inhibitors: resolving diagnostic and therapeutic dilemmas. Haemophilia. 2002 May;8(3):280-7.
18. Almagro D, Rubio R, González I, Díaz A, González A. Vida natural de los inhibidores del Factor VIII en pacientes hemofílicos. Rev Cubana Hematol Inmunol Hemoter. 1990;6:343-55.
19. Rodríguez-Merchan EC, Lee CA. Inhibitors in patients with Haemophilia. Oxford: Blackwell Science; 2002.
20. White GC. Prediction of inhibitors in hemophilia. J Thromb Haemost. 2008 Dec;6(12):2045-7.
21. Viel KR, Ameri A, Abshire TC, Iyer RV, Watts RG, Lutcher C, et al. Inhibitors of Factor VIII in black patients with hemophilia. N Engl J Med. 2009 Apr 16;360(16):1618-27. ]]>
22. Salvagno GL, Astermark J, Ekman M, Franchini M, Guidi GC, Lippi G, et al. Impact of different inhibitor reactivities with commercial factor VIII concentrates on thrombin generation. Haemophilia. 2007 Jan;13(1):51-6.
23. Kaveri SV, Dasgupta S, Andre S, Navarrete AM, Repesse´ Y, Wootla B, et al. Factor VIII inhibitors: role of von willebrand factor on the uptake of factor VIII by dendritic cells. Haemophilia. 2007 Dec;13 Suppl 5:61-4.
24. Chitlur M, Warrier I, Rajpurkar M, Lusher JM. Inhibitors in factor IX deficiency a report of the ISTH-SSC international FIX inhibitor registry (1997-2006). Haemophilia. 2009 Sep;15(5):1027-31.
]]>
25. Ter Avest PC, Fischer K, Mancuso ME, Santagostino E, Yuste VJ, Van Den Berg HM, et al. On behalf of the CANAL Study Group. Risk stratification for inhibitor development at first treatment for severe hemophilia A: a tool for clinical practice. J Thromb Haemost. 2008 Dec;6(12):2048-54.
26. Gouw SC, Ter Avest PC, Van Helden PM, Voorberg J, Van Den Berg HM. Discordant antibody response in monozygotic twins with severe haemophilia A caused by intensive treatment. Haemophilia. 2009 May;15(3):712-17.
27. Fakharzadeh SS, Kazazian HH Jr. Correlation between Factor VIII genotype and inhibitor development in hemophilia A. Semin Thromb Hemost. 2000;26(2):167-71.
28. Veltkamp JJ, Meilof J, Remmelts HG, Van der Vlerk D, Loeliger EA. Another genetic variant of haemophilia B: haemophilia B Leyden. Scand J Haemat. 1970;7(2):82-90.
29. Coyle TE, Spicer T, Michalovic D, Poiesz BJ. Moderate hemophilia B Leyden: identification by polymerase chain reaction, sequencing, and oligomer restriction. Am J Hemat. 1994 Jul;46(3):234-40.
30. Crossley M, Ludwig M, Stowell KM, De Vos P, Olek K, Brownleet G. Recovery from hemophilia B Leyden: an androgen-responsive element in the Factor IX promoter. Science. 1992 Jul 17;257(5068):377-9.
31. Picketts DJ, Mueller CR, Lillicrap D. Transcriptional control of the Factor IX gene: analysis of five Cis-acting elements and the deleterious effects of naturally occurring hemophilia B Leyden mutations. Blood. 1994 Nov 1;84(9):2992-3000. ]]>
32. Brady JN, Notley C, Cameron C, Lillicrap D. Androgen effects on factor IX expression: in-vitro and in-vivo studies in mice. Br J Haematol. 1998 May;101(2):273-9.
33. Kurachi S, Huo JS, Ameri A, Zhang K, Yoshizawa AC, Kurachi K. An age-related homeostasis mechanism is essential for spontaneous amelioration of hemophilia B Leyden. Proc Natl Acad Sci USA. 2009 May 12;106(19):7921-6.
34. De Steenwinkel F, Wesseling J, Peters M, Van Ommen Ch. Symptomatic carrier of haemophilia B Leyden: a case report. Haemophilia. 2010 Nov;16(6):965-6. doi: 10.1111/j.1365-2516.2010.02281.x.
]]>
Recibido: 13 de septiembre del 2011.
Aprobado: 11 de octubre del 2011.
]]>
Dra. Dunia Castillo-González. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800. La Habana, Cuba. Tel (537) 643 8695, 8268, Fax (537) 644 2334. Correo electrónico: ihidir@hemato.sld.cu Website: http://www.sld.cu/sitios/ihi ]]>

{kind=link}