Factores genéticos, inmunológicos y ambientales asociados a la autoinmunidad
Genetic, immunologic and environmental factors associated with autoimmunity
Lic. Sylvia Torres Odio, Ms. C. Zuzet Martínez Córdova
]]>
Hospital Clinicoquirúrgico "Hermanos Ameijeiras". La Habana, Cuba
]]>
RESUMEN
La autoinmunidad se caracteriza por una pérdida de la tolerancia inmunológica que produce la destrucción de células y tejidos propios. El sistema del complejo mayor de histocompatibilidad posee una fuerte asociación con las enfermedades autoinmunes aunque determinados genes que codifican para citoquinas y moléculas coestimuladoras incrementan la susceptibilidad genética. Estudios de concordancia entre gemelos monocigóticos demuestran el papel de los factores ambientales en la aparición de las enfermedades autoinmunes. A pesar de los avances científicos producidos en esta área de investigación, los mecanismos subyacentes de estas afecciones son desconocidos. El objetivo deeste trabajo es exponer de forma sintetizada el papel de los factores genéticos, inmunológicos y ambientales en la autoinmunidad.
Palabras clave: Enfermedades autoinmunes, sistema HLA, tolerancia central y periférica.
]]>
ABSTRACTThe autoimmunity is characterized by a loss of immunologic tolerance producing the destruction of cells and own tissues. The major complex system of histocompatibility has a close association with the autoimmune diseases although determined genes codifying for cytokines and co-stimulators molecules increase the genetic susceptibility. Concordance studies among monozygotic twins demonstrate the role of environmental factors in appearance of autoimmune diseases. Despite the scientific advances achieved in this research field, the underlying mechanisms of these affections are unknown. The objective of present paper is to expose in a summarized way the role of the genetic, immunologic and environmental factors in autoimmunity.
Key words: Autoimmune diseases, HLA system, central and peripheral tolerance.
INTRODUCCIÓN
Durante varias décadas los inmunólogos se han dedicado a descifrar el enigma de la tolerancia inmunológica. En estado fisiológico, ¿cómo los componentes del sistema inmune, en particular los linfocitos, son capaces de reaccionar contra agentes invasores (virus o bacterias) mientras permanecen inertes ante las moléculas del propio organismo? Los mecanismos de tolerancia central y tolerancia periférica son los encargados de mantener este estado de no respuesta. La pérdida o falla en alguno de ellos desencadena una respuesta inmune contra los tejidos y células del propio individuo, que se denomina autoinmunidad.1
El desarrollo de las enfermedades autoinmunes (EAs) involucra no solo la pérdida de la tolerancia, sino también factores genéticos y ambientales. La principal asociación génica se encuentra con las moléculas del HLA I-II. No obstante, la predisposición genética por sí sola no es suficiente para desencadenar una respuesta autoinmune. Las infecciones víricas y bacterianas actúan como agentes desencadenantes al crear el microambiente adecuado para la trasvasacion de las células del sistema inmune al sitio del daño. El órgano o tejido infectado libera citoquinas y proteínas capaces de alertar al sistema inmune y estimular la proliferación de clones de linfocitos T autorreactivos.2
Las EAs pueden ser órgano-específicas como la diabetes mellitus tipo 1 (DM1) y la artritis reumatoidea (AR), mientras otras son órgano-inespecíficas como el lupus eritematoso sistémico (LES).3 ]]>
Las EAs presentan un 4 % de prevalencia en los países de América del Norte y Europa, con tendencia al ascenso cada año. Este incremento está relacionado linealmente con una disminución del número de infecciones, debido a mejores condiciones socioeconómicas e higiénicas.4El objetivo de esta revisión es exponer, de forma sintetizada, aquellos factores genéticos, inmunológicos y ambientales que contribuyen al desarrollo de las EAs. La comprensión de los mecanismos inmunológicos subyacentes de estos trastornos constituye uno de los principales desafíos que debe enfrentar la inmunología.
FACTORES INMUNOLÓGICOS, GENÉTICOS Y AMBIENTALES ASOCIADOS CON LA AUTOINMUNIDAD
Factores inmunológicos
Pérdida de la tolerancia
La inducción de la tolerancia central tiene lugar en el timo para las células T inmaduras y en la medula ósea para las células B. Durante la ontogenia de los linfocitos, aquellos receptores de linfocitos T (TCR) que reconocen con elevada afinidad/avidez péptidos propios expuestos en las moléculas del HLA son eliminados por deleción clonal, con el objetivo de evitar los clones autorreactivos. Solo los clones cuyos TCR reconocen con mediana afinidad/avidez péptidos propios, maduran en los órganos linfoides secundarios. Lo anterior evidencia que las moléculas del HLA propias determinan el repertorio de TCR. Los mecanismos de tolerancia periférica incluyen la anergia clonal (ausencia de las moléculas coestimuladoras), la ignorancia y la supresión por la activación de células T reguladoras CD4+ CD25+ FOXP3+. En el reconocimiento antigénico están involucrados los segmentos a1 y b1 de las moléculas del HLA (ambos polimórficos), el péptido procesado y el TCR. De hecho, algunos péptidos procesados solo se exponen en determinadas moléculas del HLA. Por lo que las moléculas del HLA propias también determinan qué péptido puede ser reconocido por el TCR de los linfocitos T maduros. Las moléculas del HLA de un individuo determinan su respuesta inmune en dos niveles: durante la selección negativa en el timo y en la selección de los péptidos en la periferia.5
Las nuevas líneas de investigación de las EAs se enfocan hacia los genes que codifican para moléculas implicadas en la inducción de la tolerancia central y periférica. Estos genes se encuentran en cualquier cromosoma y codifican para proteínas implicadas en la selección de los linfocitos y moléculas que actúan como receptores de muerte o moléculas coestimuladoras. La mayoría de las EAs son poligénicas lo que dificulta el conocimiento del agente desencadenante. Las EAs provocadas por una mutación en un único gen (monogénicas), las cuales son poco comunes, proveen evidencias clínicas y experimentales de la contribución de los diferentes mecanismos de control de la autorreactividad.5
Factores genéticos
Asociación con el sistema mayor de histocompatibilidad ]]>
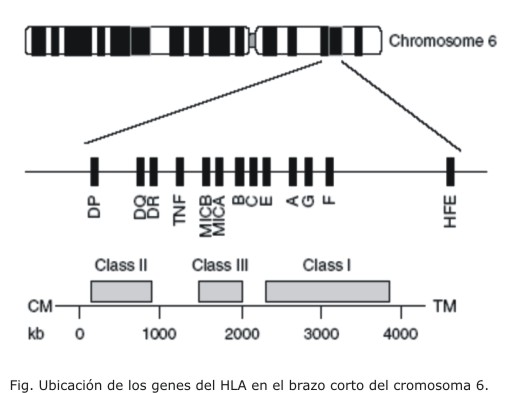
Los genes que codifican para las moléculas del HLA se ubican en el brazo corto del cromosoma 6 en la región del complejo mayor de histocompatibilidad (MHC). Estos genes presentan herencia autosómica, expresión codominante y codifican para las moléculas de clase I, II y III (fig.).6En los mamíferos las moléculas HLA clase I (HLA-I) codificadas por los genes HLA-A, B, C, E, F, G se expresan en todas las células nucleadas y en las plaquetas. Las moléculas HLA clase II (HLA-II) son productos de los genes HLA-DP, DQ, DR, DM, DO y se expresan constitutivamente en los linfocitos B, los monocitos, macrófagos, células dendríticas, células endoteliales, células epiteliales intestinales, células hematopoyéticas tempranas y en linfocitos T activados. La región de clase III denominada HLA-no clásico contiene una colección de aproximadamente 20 genes. En esta región se incluyen aquellos que codifican para las proteínas del complemento, componentes involucrados en el procesamiento intracelular de péptidos (TAP1, TAP2) y moléculas de expresión en la superficie de células epiteliales (MICA-MICB).7
La función fundamental de las moléculas HLA-I y HLA-II es unir péptidos propios y extraños con el objetivo de transportarlos hacia la membrana celular. Una vez expuestos son reconocidos por el TCR, por lo que tienen un papel central en la ejecución de la respuesta inmune. Las moléculas HLA-I presentan fundamentalmente péptidos citosólicos (como los virales o tumorales) a las células T citotóxicas CD8+ mientras que las moléculas HLA de clase II presentan, generalmente, péptidos extracelulares (como los bacterianos) a los linfocitos T cooperadores CD4+. Esta división funcional de la presentación de los péptidos asegura la activación de las células T (CD8+ y CD4+) y por consiguiente, la respuesta inmune adecuada para cada tipo de antígeno.8
El sistema del HLA posee dos propiedades fundamentales que dificultan la comprensión acerca de los genes implicados en la predisposición a las EAs: el polimorfismo y el desequilibrio de ligamiento (LD).9
Las moléculas I-II son las más polimórficas de todo el genoma. Esta propiedad determina que para cada loci existen múltiples alelos cuyas secuencias de ADN solo difieren en unos pocos nucleótidos. Estas mutaciones locales se conocen como polimorfismo de simple nucleótido (NSP de sus siglas en inglés). En enero de 2010, la IMGT/HLA Database reportó un total de 4 447 alelos: 3 249 del HLA clase I y 1 198 del HLA clase II. Pese a esta diversidad, un individuo presenta solamente 2 alelos. La combinación de los genes del HLA representa un haplotipo que es completamente heredado de los padres y fenotípicamente expresado. Se estima que más de 100 millones de fenotipos diferentes resultan de todas las combinaciones posibles del sistema HLA. El haplotipo de un individuo es único y lo hace un marcador ideal para estudios genéticos. El extenso polimorfismo de las moléculas del HLA asegura que existan individuos con óptimos genotipos que respondan contra los antígenos no propios. Esta pudiera ser la razón por la cual se asocian determinadas infecciones con uno o varios alelos en específico.9, 10
Los genes ubicados en la región del MHC presentan una alta asociación génica. Esta propiedad se conoce como desequilibrio de ligamiento (DL) (en inglés linkage desequilibrium) y describe la tendencia de determinados genes a heredarse juntos dada su cercanía. Lo anterior determina que la frecuencia de estos genes (en un simple haplotipo) en la población es mayor que su herencia individual. El mejor ejemplo de DL lo ofrece el haplotipo HLA A1-B8-DR3-DQ2, conocido como el haplotipo autoinmune, con una frecuencia de aparición del 10 % en las poblaciones del norte de Europa. En la población noruega se esperaría una frecuencia de este haplotipo del 0,3 % cuando realmente es del 7,7 % debido al DL. En las moléculas HLA-II este fenómeno es más pronunciado, principalmente en las moléculas HLA-DR y HLA-DQ. La presencia de alelos específicos del HLA-DR tiene un valor predictivo sobre el alelo HLA-DQ con un alto nivel de certeza. Esta propiedad hace difícil determinar qué genes dentro del MHC son los que contribuyen de forma primaria y secundaria en la predisposición a la enfermedad.10,11
La base genética de las EAs surge del estudio de individuos de una misma familia. En la DM1, la concordancia entre gemelos monocigóticos (idéntico ADN) es de 30-70 % lo que indica la existencia de una predisposición genética. Si bien este valor no es del 100 %, indica el papel que desempeñan los agentes ambientales e inmunológicos en contribuir a la enfermedad en aquellos individuos con una base genética. Dentro de la genética de las EAs, la mayor asociación se encuentra con las moléculas del HLA. La concordancia para hermanos con idéntico HLA es del 15 % comparada con el 1 % para hermanos con un HLA no idéntico. Esta cifra, así como la tabla 1, son indicativas de la fuerte asociación existente entre las moléculas del HLA y la predisposición a desarrollar una enfermedad autoinmune. En algunas enfermedades esta asociación es más fuerte como en la espondilitis anquilosante (EA), mientras en otras es más débil como en la miastenia gravis (MG).12
Genes del HLA-no clásico
]]> Estudios en pacientes con DM1, LES y enfermedad celíaca (EC) revelan la presencia de un gen en la región telomérica del HLA-I "marcado con el microsatélite D6S2223", involucrado en la predisposición a estas enfermedades. Un candidato posible dentro de esta zona es el gen que codifica para una serinoproteasa timoespecífica (PRSS16), la cual se especula está involucrada en la selección positiva de los clones de células T.13
Genes independientes del MHC asociados con las EAs
AIRE
El síndrome de poliendocrinopatía tipo 1 (SPE1) es causado por una mutación en el gen AIRE (autoimmune regulator) (tabla 2). Las personas afectadas con el SPE1 presentan tres condiciones: la enfermedad de Addison, hipoparatiroidismo y candidiasis mucocutánea. Estos individuos presentan anticuerpos contra una gran variedad de antígenos periféricos lo que implica afectaciones a otros órganos que incluyen: mala absorción, alopecia, falla gonadal y hepatitis crónica activa.14,15
El gen AIRE se encuentra en el brazo corto del cromosoma 21, el cual codifica para la proteína AIRE (pAIRE) que actúa como un factor de transcripción "no clásico". La pAIRE regula la transcripción de antígenos propios órgano-específicos en las células epiteliales del timo (CET) por lo que tiene un importante papel en la tolerancia central. Las mutaciones de este gen conducen a la selección positiva de linfocitos cuyos TCR reconocen antígenos propios. La subunidad a del receptor muscular de acetil-colina, el autoantígeno de la miastenia gravis, es codificado por un gen cuya expresión depende del gen AIRE en las CET. Las hipótesis sobre el papel del gen AIRE en el mecanismo fisiopatológico de las EAs incluyen: la organización del estroma tímico, el control de la tolerancia de los timocitos, la regulación de la respuesta de las células B y T ante la presentación antigénica y la diferenciación a células T reguladoras (Treg) CD4+CD25+ FOXP3+.16
CTLA4
La molécula CTLA4 (cytotoxic T lymphocyte antigen 4) (tabla 2) es un receptor inhibidor expresado en los linfocitos T cuyos ligandos son las moléculas coestimuladoras CD80-CD86. La interacción CD80-CTLA4 induce la anergia del linfocito, lo que constituye un mecanismo de tolerancia periférica. El modelo animal carente de esta molécula desarrolla un síndrome fatal de infiltración linfocitiaria. Los polimorfismos que disminuyen la expresión y/o función de esta molécula pueden conducir a una exagerada activación de linfocitos. La molécula CTLA4 se asocia con enfermedades mediadas por células B (enfermedad de Graves) así como aquellas mediadas por células T (DM1). No obstante, el riesgo relativo asociado es bajo lo cual indica la participación de otros genes en el desarrollo de la autoinmunidad. 17
FOXP3
El FOXP3 es un factor de transcripción de la familia de forkhead producido en altos niveles por las células Treg CD4+CD25+ FOXP3+ (tabla 2). Estas células están implicadas en los mecanismos de inducción de tolerancia periférica. El modelo animal carente del gen que codifica para este factor, desarrolla una EA sistémica implicada con la deficiencia de células Treg conocida como IPEX (pérdida de la regulación inmune, poliendocrinopatía, enteropatía y síndrome del crom. X).18-20
]]> Fas/FasLLas moléculas FasL/CD178 y Fas/CD95 (tabla 2) son proteínas transmembrana que pertenecen a la familia del factor de necrosis tumoral (FNT) y al receptor del FNT, respectivamente. El sistema Fas/FasL induce la muerte celular por apoptosis, de forma que elimina los clones de las células T y B autorreactivas que median la autoinmunidad. Las mutaciones en los genes que codifican para estas moléculas inducen un síndrome autoinmune linfoproliferativo que se caracteriza por linfoproliferación, manifestaciones autoinmunes y aumento del TCR áâ en linfocitos T CD4-CD8-..21,22
Factores ambientales
Los valores de concordancia entre gemelos monocigóticos son indicativos del papel de los factores ambientales en el desarrollo de la autoinmunidad. Dentro de este grupo se encuentran las infecciones (virus, parásitos, bacterias, hongos), las hormonas y la pérdida de la regulación del sistema inmune. El mecanismo de acción propuesto para estos factores se basa en la liberación de sustancias proinflamatorias que inducen la expresión de señales de peligro y la consecuente activación de clones de linfocitos T autorreactivos.23
Agentes infecciosos
Las infecciones están implicadas en la inducción y en la protección a las EAs en individuos genéticamente predispuestos. La compresión del mecanismo subyacente de este papel dual, ofrece nuevas formas de control y tratamiento de estas enfermedades.24
Papel como agentes desencadenantes
Su rol como agentes detonantes se ha visto en la AR y Proteus mirabilis, EA y Klebsiella pneumoniae, DM1 y Coxsackievirus. Las hipótesis que explican el mecanismo de acción incluyen: el mimetismo molecular, el reconocimiento dual por parte de los TCRs y el aumento del procesamiento y presentación antigénica de autoantígenos durante la infección.24
La base del mimetismo molecular radica en la similitud de secuencia que comparten péptidos propios y péptidos virales. Este mecanismo fue demostrado por primera vez al inmunizar ratones con la polimerasa del virus de la hepatitis B. Esta enzima comparte 6 aminoácidos con la proteína básica de la mielina. Después de ser infectado, el animal desarrolla lesiones inflamatorias en el sistema nervioso central debido a la activación de linfocitos T autorreactivos.25
Los TCRs de los linfocitos T reconocen diferentes péptidos en el surco de la molécula del HLA siempre que mantengan la misma distribución de cargas y la orientación espacial. De ahí que moléculas propias y extrañas que presenten esta similitud sean reconocidas por los linfocitos y produzcan una respuesta inmune.25 ]]>
La creación de un microambiente inflamatorio incrementa el procesamiento y presentación de antígenos propios producto del daño tisular y la expresión de moléculas coestimuladoras. En este medio los linfocitos T anergizados pueden activarse y estimular la respuesta inmune contra antígenos propios.26Las infecciones pueden, además, modificar las manifestaciones clínicas asociadas a una EA. Un estudio de cohorte realizado en pacientes con el síndrome de Sjögren indicó una prevalencia del 3 % del virus de la hepatitis C. Los pacientes infectados presentan mayor fotosensibilidad y número de crioglobulinas "no asociado a los síntomas clásicos de la crioglobulinemia", al compararlos con aquellos que no están infectados.26
Papel como agentes protectores
Paradójicamente, los agentes infecciosos también pueden suprimir el desarrollo de una EA. Existe una correlación inversa entre la prevalencia de las EAs y las infecciones en los países industrializados, principalmente en Europa y América del Norte. La causa principal de este comportamiento radica en las mejores condiciones higiénico-sanitarias, socioeconómicas y la elevada utilización de antibióticos y vacunas. Hasta la actualidad, no se ha encontrado una causa genética que explique esta conducta. Prueba de ello es que la frecuencia del LES es extremadamente baja en África comparada con la población negra americana, aun cuando ambas poblaciones derivan del mismo grupo étnico. Las bases fisiológicas plantean que los agentes infecciosos inducen la liberación IL-10 y TGF-B por parte de las células T reguladoras CD4+ CD25+ FOXP3+, estas interleucinas inhiben la respuesta Th1 y Th2; como consecuencia ante la ausencia de infecciones se elimina este mecanismo de regulación de la respuesta inmune.27,28
De forma general, no todos los factores implicados en la evolución de las EAs contribuyen de igual manera. En la espondilitis anquilosante, el riesgo relativo (RR) asociado al antígeno HLA-B27 es elevado (mayor de 100 %) sin embargo, el papel que desempeñan los agentes inmunológicos y ambientales permanece sin dilucidar. Por otra parte, en la enfermedad celíaca el agente desencadenante (gluten) y el mecanismo inmunológico son conocidos, sin embargo, el RR asociado a los antígenos HLA-DQ2/DQ8 es menor (aproximadamente 30 %).10
La elevada incidencia y las complicaciones sistémicas que acarrean estas enfermedades estimulan nuevas investigaciones acerca de los genes de predisposición y los agentes desencadenantes. Los avances científicos logrados en este campo posibilitan el desarrollo de métodos de tratamiento y prevención más eficaces. A pesar de los conocimientos alcanzados en esta área de investigación, la multitud de factores implicados en la aparición de las EAs constituye un reto que debe enfrentar la inmunología moderna.
REFERENCIAS BIBLIOGRÁFICAS
1. Adams DD, Knight JG, Ebringer A. Autoimmune diseases: Solution of the environmental, immunological and genetic components with principles for immunotherapy and transplantation. Autoimmunity Rev. 2010;9:525-30. ]]>
2. Ermann J, Fathman CG. Autoimmune diseases: genes, bugs and failed regulation. Nature Immunology. 2001;2:759-62.
3. Abbas K, Litcham H, Pillai S. Inmunología celular y molecular. Sexta edicion. Elsevier Saunders; 2010.
4. Browning M, McMichael A, ed. HLA and MHC: genes, molecules and function. Oxford: BIOS Scientific Publ. Ltd.; 1996.
5. Arnold B. Levels of peripheral T cell tolerance. Transplant Immunol. 2002;10:109-14.
6. Undlien DE, Lie BA, Thorsby E. HLA complex genes in type 1 diabetes and other autoimmune diseases. Which genes are involved? Trends Genet. 2001;17:93-100. ]]>
7. Thorsby E, Lie BA. HLA associated genetic predisposition to autoimmune diseases: Genes involved and possible mechanisms. Transplant Immunol. 2005;14:175-82.
8. Thorsby E, Lie BA. Several genes in the extended human MHC contribute to predisposition to autoimmune diseases. Curr Opin in Immun. 2005;17:526-31.
9. Cassinotti A, Birindelli S, Clerici M, Trabattoni D, Lazzaroni M, Ardizzone S, et al. HLA and Autoimmune Digestive Disease: A Clinically Oriented Review for Gastroenterologists. Am J Gastroenterol. 2009;104:195-217.
10. Caillat-Zucman S. Molecular mechanisms of HLA association with autoimmune diseases. Tissue Antigens. 2008;73:1-8.
11. Shiina T, Inoko H, Kulski JK. An update of the HLA genomic region, locus information and disease associations. Tissue Antigens. 2004;64:631-49. ]]>
12. Waldner H. The role of innate immune responses in autoimmune disease development. Autoimmunity Rev. 2009;8:400-04.
13. Lie BA, Akselsen HE, Bowlus CL, Gruen JR, Thorsby E, Undlien DE. Polymorphisms in the gene encoding thymus-specific serine protease in the extended HLA complex: a potential candidate gene for autoimmune and HLA-associated diseases. Genes Immun. 2002;3:306-12.
14. Björses P, Aaltonen J, Horelli-Kuitunen N, Yaspo M, Peltonen L. Gene defect behind APECED: a new clue to autoimmunity. Hum Mol Gen. 1998;7:1547-53.
15. Fierabracci A. Recent insights into the role and molecular mechanisms of the autoimmune regulator (AIRE) gene in autoimmunity. Autoimmun Rev. 2011;10:137-43.
16. Rioux JD, Abbas AK. Paths to understanding the genetic basis of autoimmune disease. Nature. 2005;435:584-89. ]]>
17. Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506-11.
18. Langier S, Sade K, Shmuel K. Regulatory T cells: The suppressor arm of the immune system. Autoimmun Rev. 2010;10:112-15.
19. Fontenot JD, Gavin MA, Rudensky AY. FOXP3 programs the development and function of CD4+CD25+ regulatory T cells. Nature Immunol. 2003;4:330-36.
20. Bennett CL. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nature Genet. 2001;27:20-21.
21. Fisher GH. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935-46. ]]>
22. Atkinson JP. Complement deficiency: predisposing factor to autoimmune syndromes. Clin Exp Rheumatol. 1989;7:95-101.
23. Zanelli E, Breedveld FC, de Vries RRP. HLA association with autoimmune disease: a failure to protect? Rheumatology. 2000;39:1060-66.
24. Proal AD, Albert PJ, Marshall T. Autoimmune disease in the era of the metagenome. Autoimmunity Rev. 2009;8:677-81.
25. Doria A, Sarzi-Puttini P, Shoenfeld Y. Infections, rheumatism and autoimmunity: the con.icting relationship between humans and their environment. Autoimmun Rev. 2008;3:243-46.
26. Ryan KR, Patel SD, Stephens LA, Anderton SM. Death, adaptation and regulation: the three pillars of immune tolerance restrict the risk of autoimmune disease caused by molecular mimicry. J Autoimmun. 2007;29:262-71. ]]>
27. Bach JF. Infections and autoimmune diseases. J Autoimmun. 2005;25:74-80.
28. Bach JF. The effect of infections on susceptibility to autoimmune and allergic disease. N Engl J Med. 2002;347:911-20.
Recibido: 22 de febrero de 2011. ]]> Aprobado: 12 de marzo de 2011.
Lic. Silvia Torres Odio. Hospital Clinicoquirúrgico "Hermanos Ameijeiras". San Lázaro 701, esquina a Belascoaín. Centro Habana, La Habana, Cuba. CP 10700. Correo electrónico: sylviat@infomed.sld.cu ]]>
{kind=link}
{kind=link}