
Complejo Científico Ortopédico Internacional "Frank País"
Dr. Rolando Gondres Argote,1 Dra. Daysi Socorro Febles,2 Dra. Osana Vilma Rondón García3 y Dra. Martha Melo Víctores1
Gondres Argote R, Socorro Febles D, Rondón García OV, Melo Víctores M. Síndrome de Holt y Oram. Presentación de un caso. Rev Cubana Ortop Traumatol 2000;14(1-2):56-61
Se presentó un caso de anomalías musculoesqueléticas y cardiopatía congénita con acortamiento del miembro superior izquierdo y CIA, ausencia de pulgares en ambas manos asociado a hombros estrechos tipo foramen oval. Se realizaron exámenes complementarios que confirman el diagnóstico clínico. En el síndrome de Holt y Oram se señala herencia autosómica dominante y al no encontrar antecedentes familiares en nuestro caso, se planteó una mutación genética como causa de la aparición aislada de este síndrome.
DeCS: ANOMALIAS MUSCULOESQUELETICAS; CARDIOPATIAS CONGENITAS; PULGAR/anomalías; MANO/anomalías; ANTEBRAZO/anomalías; HOMBRO/anomalías; NIÑO.
El síndrome fue descrito por primera vez por Holt y Oram en el año 1960 en un niño que presentaba defecto septal interauricular, anomalías del pulgar y arritmia supraventricular.1,2
Desde entonces han sido publicados otros casos con cuadros similares.3-9 Hasta el año 1967 se habían publicado 30 casos.10
En Cuba, en el año 1977, fue publicado por Savio y otros el caso de un niño de 4 años que presentaba acortamiento del miembro superior izquierdo, agenesia del pulgar y tetralogía de Fallot.11
En la década del 80 Goldstein y Brown señalaron que, aunque no se había definido la acción bioquímica del gen mutante, se suponía que actuaba rompiendo el proceso embrionario, importante para la formación de las extremidades superiores y el corazón.12
]]> En publicaciones internacionales recientes han sido reportados otros casos13-18 y diferentes autores coinciden en señalar mutación en el gen TBX5, factor de transcripción como causa en el humano de las malformaciones que se observan en este síndrome y muchas mutaciones han sido identificadas.19-22Anomalías que puede presentar el síndrome de Holt y Oram
Anomalías del esqueleto
Diversos grados de malformación en el miembro superior y hombro; desde hipoplasia de dedos hasta focomelia. Es más frecuente en lado izquierdo, en proporción de 2:1. No hay correlación entre la severidad del defecto del miembro y el defecto cardíaco.
Las anomalías del pulgar son frecuentemente bilaterales, pero asimétricas; son habituales los tipos de ausencia digital, presencia de remedo digital o de trifalangismo. Uno o los tres segmentos del primer rayo pueden estar duplicados. El 1er. metarcapiano suele estar alargado y delgado o ausente; puede haber sindactilia.
Las anomalías del carpo adoptan la forma de persistencia de la primitiva fila central del carpo, con un hueso accesorio del lado cubital del escafoide.
Poznanski y otros encontraron que los huesos carpianos adicionales constituían la anomalía más chocante del esqueleto en sus pacientes.23
Puede existir rotación de la escápula, anormalidad de la articulación acromioclavicular, deformidad de la clavícula, de la epitroclea humeral y del esternón, luxación de la cabeza del radio o ausencia del radio, defectos del cúbito y agenesia del sacro.2,24,25
Anomalías cardiovasculares
El defecto auricular septal, a veces con arritmia, siendo el osteum secundum el defecto interauricular más frecuente y el osteum primus excepcional. El defecto septal ventricular ha sido también el defecto más común. Otros defectos han sido la transposición de grandes vasos y el origen anómalo de coronaria izquierda. Cerca de un tercio de los pacientes tienen otro tipo de defecto cardíaco congénito.2
]]> Anomalías ocasionalesHipertelorismo, presencia de ductus arterioso, estenosis pulmonar, ausencia del músculo pectoral mayor, pectus escavatum, escoliosis, anomalías vertebrales, ausencia de uno o más centros de osificación en la muñeca.2
Etiología
Autosómica dominante con expresión variable, tendiente a ser más severa en el sexo femenino. Hay casos que pueden presentar mutaciones esporádicas del gen.2,12,19-22,26
Presentación de un caso
Al examen clínico (fig. 1)
Fig. 1. Características externas.
]]> Exámenes complementarios (figs. 2 y 3).Radiología:

Fig. 2. Miembro superior derecho.
]]>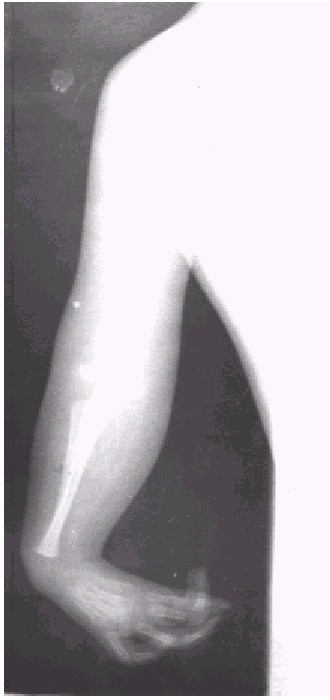
Fig. 3. Miembro superior izquierdo.
No se visualiza núcleos de asificación en codos y manos.
Telecardiograma:

Fig. 4. Telecardiograma.
]]>
Fig. 5. Electrocardiograma. Derivación V1.
Ecocardiograma:
CIA, tipo foramen oval, 11 mm (fig. 6). Insuficiencia mitral y tricuspidea ligera.
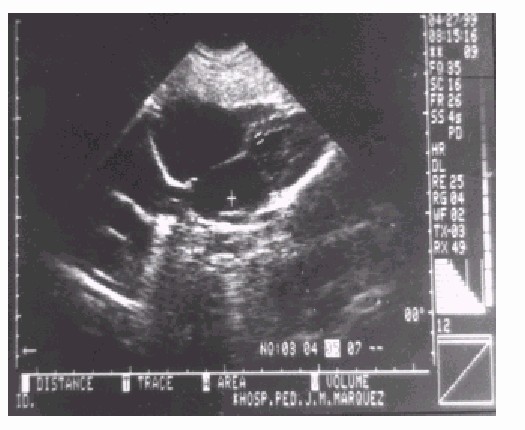
Fig. 6. Ecocardiograma.
El desarrollo embriológico del tubo cardíaco y de las extremidades superiores se inicia hacia la 4ta. semana de vida embrionaria. Esta relación puede explicar la asociación de cardiopatías con malformación de miembros superiores.
En nuestro caso se cumplen los criterios para el diagnóstico del síndrome de Holt y Oram.
]]> No pudimos encontrar antecedentes familiares en este caso, por lo que planteamos la posibilidad de una mutación genética como explicación a la aparición aislada del síndrome en un solo miembro de la familia.A case of musculoskeletal abnormalities and congenital heart disease with shortness of the left upper limb and interauricular communication (IAC), and absence of thumbs in both hands associated with foramen ovale-like narrow shoulders was presented. The clinical diagnosis was confirmed by complementary tests. Dominant autosomal inheritance is stressed in the Holt and Orams syndrome and, as no familiy history was found in our case, a genetic mutation was considered as the cause of the isolated appearance of this syndrome.
Subject headings: MUSCULOSKELETAL ABNORMALITIES; HEART DEFECTS; CONGENITAL; THUMB/abnormalities; HAND/abnormalities; FOREARM/abnormalities; SHOULDER/abnormalities; CHILD.
Un cas danomalies musculo-squelettiques et de cardiopathie congénitale avec raccourcissemet du membre supérieur gauche et CIA, ainsi quabsence de tous les deux pouces associée à des épaules étroits type trou ovale, a été présenté. Des examens complémentaires confirmant le diagnostique clinique ont été réalisés. Dans le syndrome de Holt et Oram, une hérédité autosomique dominante fut signalée, et étant donné quon na pas rencontré dantécédents familiaux dans notre cas, une mutation génétique comme cause de survenue isolée de ce syndrome fut établie.
Mots clés: ANOMALIES MUSCULO-SQUELETTIQUES; CARDIOPATHIES CONGÉNITALES; POUCE/anomalies; MAIN/anomalies; AVANT-BRAS/anomalies; ÉPAULE/anomalies.
1. Holt M, Oram S. Familial heart disease with skeletal malformation. Br J 1960;(22):236.
2. Smith DW, Lyon JK. Recognizable patterns of human malformation genetic, embriologic and clinical aspects. 3ra. ed. Phyladelphia; WB Saunders, 1982;232.
3. Kuhn E. A primary pulmonary hypertension, congenital heart disease and skeletal anomalies in three generations. Jap Heart J 1963;(4):205.
4. Homes LB. Congenital heart disease and upper-extremity deformities. New Engl J Med 1965;272:437.
5. Gall JC. Holt Oram Syndrome clinical and genetic study of a large family. Am J Hum Genet 1965;(18):187.
6. Pruzanki W. Familial congenital malformations of the heart and upper limbs a syndrome of Holt Oram. Cardiology 1964;(45):21.
7. Sánchez CA. Holt Oram Syndrome. Acta Paediatr Scand 1967;(56):313.
8. Ehlers KF, Engel MA. Familial congenital heart disease: genetic and environmetal factors. Circulation 1966;(34):503.
9. Solit RW. Congenital heart disease and upper extremity defects: a case report (Holt Oram Syndrome). Cardiovasc Surg 1976;(14):76.
10. Chang CH. Holt Oram Syndrome. Radiology 1967;(88):479.
11. Savio BA. Syndrome de Holt y Oram: presentación de un caso. Rev Cubana Pediatr 1977;49(2):193.
12. Goldstein JL, Brown MS. Genética y enfermedades vasculares, en: Braunwald E. Tratado de Cardiología. La Habana: Editorial Pueblo y Educación, 1981;3(2):1855.
13. Basson CT. Holt Oram Syndrome vs heart-hand syndrome. Circulation; 2000 May 9;101(18):E191.
14. Kranidis A; Filippatos G; Karvounis H. A case of Holt Oram Syndrome. Int J Cardiol; 2000 Mar 31;73(1):95-6.
15. Tongsong T; Chanprapaph P. Prenatal sonographic diagnosis of Holt Oram Syndrome. J Clinic Ultrasound; 200 Feb;28(2):98-100.
]]>16. Tate DE, Gupta A, Kleinert HE. Bipartite scaphoid with proximal pole osteonecrosis in a patient with Holt Oram syndrome J Hand Surg (Br);2000 Feb;25(1): 112-4.
17. Frota F, Pereira W, Leiria TL, Vallenas M, Blacher C. Holt-Oram Syndrome revisited. Two patients int the same family. Arq Bras Cardiol; 1999 Nov;73(5):429-34.
18. Brockhoff CJ, Kober H, Tsilimingas N, Dapper F, Myunzel T, Holt-Oram Syndrome. Circulation; 1999 Mar;99(10): 1395-6.
19. Hatcher CJ, Goldstein MM, Mah CS, Delia CS, Basson CT. Identification and localization of TBX5 transcription factor during human cardiac morphogenesis. Dev Dyn;2000 Sep;219(1):90-5.
20. Yang J, Hu D, Xia J, Yang Y, Yin B, Hu J, Zhou X. TBX5 mutation in Chinese patients with Holt-Oram syndrome. Chung Hua I Hsueh I Chuan Hsueh Tsa Chih; 2000 Aug, 17(4):233-5.
21. Liberatore CM, Searcy-Schirck RD, Yutzey KE. Ventricular expression of TBX5 inhibits normal heart hamber development. Dev Biol; 2000 Jul;223(1):169-80.
22. Yang J, Hu D, Xia J, Yang Y, Ying B, Hu J, Zhou X. Three novel TBX5 mutations in Chinese patients with Holt-Oram syndrome. Am J Med Genet; 2000 Jun;92(4):237-40.
23. Poznanski AK, Gall TC, Stern AM. Skeletal manifestations of the Holt Oram Syndrome. Radiology 1970, 94:45.
24. Tachdjian MO, MB Ortopedia Pediátrica. 2da. ed. México: Interamericana. 1994, vol:205.
25. Ozonoff MB. Radiología en Ortopedia Pediátrica. Buenos Aires. Ed. Médica Panamericana, 1982:113-5.
]]>26. Reston P. Congenital skeletal anomalies; skeletal dysplasias, chromosomal disorders. En: Sutton D. A textbook of radiology and imaging. 5ta. ed. Edinburgh: Churchil Livingstons, 1993:3.
Recibido: 26 de abril del 2000. Aprobado: 18 de julio del 2000.
Dr. Rolando Gondres Argote. Complejo Científico Ortopédico Internacional "Frank País". Avenida 51 No. 19603 e/ 196 y 202, La Lisa, Ciudad de La Habana, Cuba.
1 Especialista de I Grado en Pediatría. Jefe de Servicio de Pediatría.
2 Especialista de II Grado en Pediatría.
3 Especialista de I Grado en Radiología. Jefe del Departamento de Radiología.