Anomalías del desarrollo del segmento anterior
Anomalies in the development of the anterior segment
Dra. Lourdes R Hernández Santos, Dr. Pedro Daniel Castro, Dra. Lucy Pons Castro, Dra. Rosa M Naranjo Fernández, Dra. Milagros Dorrego Oduardo, Dr. Alejandro Arias Díaz.
Instituto Cubano de Oftalmología "Ramón Pando Ferrer". La Habana, Cuba.
]]>
RESUMEN
Las disgenesias del segmento anterior forman parte de un grupo de anomalías del desarrollo ocular que comparten características clínicas y se acompañan en su gran mayoría de glaucoma. En esta revisión se empleó la clasificación usada por Taylor, que las divide en anomalías originadas en las células de la cresta neural, anomalías ectodérmicas y anomalías de origen global, se describen sus características clínicas, tratamiento y alteraciones genéticas. Debido a la variabilidad de cuadros clínicos, se hace más necesaria la identificación de los genes en el desarrollo del segmento anterior que permite la clasificación de estas disgenesias, según la mutación genética subyacente.
Palabras clave: disgenesias del segmento anterior, cresta neural, anomalías ectodérmicas, mutaciones genéticas.
ABSTRACT
The anterior segment dysgeneses are part of a group of anomalies of the ocular development that share clinical characteristics and are mostly accompanied by glaucoma. This article used Taylor´s classification, which divides them into anomalies originated in the the neural crest cells , ectodermic anomalies and anomalies of global origin, describing their clinical characteristics, treatment and genetic alterations. Taking into account the diversity of clinical findings, it is increasingly important to identify gens in the development of the anterior segment which allow the classification of these dysgeneses according to the underlying genetic mutation.
Key words: anterior segment dysgeneses, neural crest, ectodermic anomalies, genetic mutations.
INTRODUCCIÓN ]]>
El espectro de anomalías del desarrollo conocido como disgenesias del segmento anterior, algunas veces llamadas disgenesias mesenquimatosas, fueron agrupadas antiguamente como síndrome de clivaje del segmento anterior.1 Estas constituyen un grupo de enfermedades definidas como anomalías del desarrollo que muestran algunas características comunes y una alta prevalencia de glaucoma asociado.Estas son disgenesias que se ven con relativa frecuencia en consulta. Su conocimiento permite una mejor orientación ante estos pacientes, los cuales presentan complicaciones asociadas con pronósticos desfavorables en su mayoría al no ser diagnosticadas precozmente.
Existen numerosas clasificaciones, algunos autores la clasifican desde el punto de vista embriológico en anomalías del desarrollo derivadas del cresta neural (ASDnc) y no derivadas de la cresta neural (ASDnon nc).2 Otros autores las dividen en periféricas, centrales y una combinación de ambas.3-6 También está la clasificación usada por Taylor,7 más usada por considerarse más abarcadora:
1. Anomalías originadas en las células de la cresta neural.
2. Anomalías ectodérmicas.
3. Anomalías de origen global.
En esta revisión se agrupan de forma organizada estas anomalías. Esto permite que el personal médico pueda orientar con más precisión su diagnóstico, conocer el cuadro clínico y brindar un tratamiento individualizado para cada paciente. También se relacionan las anomalías con sus alteraciones genéticas, que aunque mucho se ha avanzado en los últimos años, el conocimiento actual sobre las alteraciones genéticas en estas disgenesias es incompleto y quedan algunas condiciones pendientes de concluir su condición genética.
SOBRE EMBRIOLOGÍA OCULAR
]]> Estas anomalías ocurren con un gran número de alteraciones oculares y sistémicas, pero hay que tener presente que en algunos pacientes se pueden presentar de forma aislada como detallamos en esta revisión. Se revisaron artículos de revistas, artículos de internet y libros sobre esta temática desde enero a diciembre del 2010.Para entender estas anomalías es importante conocer la embriología del ojo. De esta recordaremos el origen de las diferentes estructuras del segmento anterior del globo ocular.2,3
Origen embriológico de las estructuras oculares:
- Ectodermo superficial:
]]> - Neuroectodermo:· Epitelio conjuntival.
· Epitelio corneal.
· Cristalino.
· Glándula lagrimal y sistema de drenaje.
· Epitelio, glándulas y cilios de la piel de párpados y carúncula.
· Músculo esfínter y dilatador del iris.
· Epitelio pigmentario del iris, retina y cuerpo ciliar.
· Epitelio ciliar no pigmentado.
· Zónula del iris.
· Retina neurosensorial.
· Humor vítreo.
· Nervio óptico.
- Células de la cresta neural craneal:
]]>· Estroma y endotelio corneal.
· Esclera (excepto porción temporal).
· Malla trabecular.
· Tejido conectivo del iris.
· Canal de Schlemm.
· Ángulo camerular
· Estroma de coroides.
· Músculo del cuerpo ciliar.
· Melanocitos (uveales y epiteliales). ]]>
- Mesodermo:
· Fibras de los músculos extraoculares.
· Humor vítreo.
· Porción temporal de la esclera.
· Revestimiento endotelial de los vasos sanguíneos. ]]>
ANOMALÍAS ORIGINADAS EN LAS CÉLULAS DE LA CRESTA NEURAL
Embriotoxon posterior.
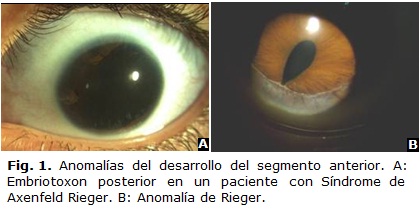
Es un desplazamiento anterior prominente de la línea de Schwalbe. Es visible con lámpara de hendidura como una línea blanca irregular concéntrica al limbo.2,6,7 Gonioscópicamente aparece como una cresta continua o rota que protruye en la cámara anterior y puede tener pigmentos en su superficie interna. Aparece aislada en 8 a 15 % de los pacientes normales o puede acompañar al síndrome de Axenfeld-Rieger (Fig. 1 A) o al síndrome de Alagille (displasia arteriohepática). No tiene un origen genético definido. Los casos aislados no requieren tratamiento.
Hipoplasia del iris / Iridogoniodisgenesia.
La hipoplasia del estroma del iris puede aparecer aislada o asociada a disgenesias del ángulo camerular. El iris de color gris o carmelita representa el epitelio pigmentario de este visto a través del estroma del iris hipoplásico. El glaucoma que aparece se debe a goniodisgenesias, el ángulo es normal por gonioscopía y tiene poca respuesta a la goniotomía.6,7 Una característica del iris que se pasa por alto y es patognomónico es que el collarete del iris está ausente, o pequeño y periférico con hebras del iris atadas a la línea de Schwalbe como en el síndrome de Axenfeld-Rieger.
Tiene dos locus genéticos involucrados que son el 6p25 y el 4q25 (FOXC1, PITX2). Hay otros reportes de afectación del gen FOXE3 (locus 1p32).2,8 Algunos autores lo han asociado a anomalías sistémicas como hipoplasia maxilar, anomalías dentarias, piel periumbilical redundante e hispopadias.
Síndrome de Axenfeld-Rieger.
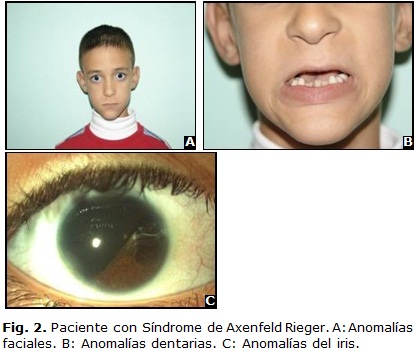
]]> Fue descrito en 1920 por Axenfeld como anomalía de Axenfeld, donde aparecía un embriotoxon posterior y bandas que iban de la periferia del iris al embriotoxon.2,9,10 Más tarde en 1934 Rieger describió lo mismo pero acompañado de anomalías del iris como corectopia y policoria, y lo reconoció como anomalía de Rieger (Fig. 1 B). Al asociarse ambas se designa como anomalía de Axenfeld-Rieger.Esta última cuando se asocia a manifestaciones sistémicas se denomina síndrome de Axenfeld-Rieger (SAR) y es el término que se prefiere en la actualidad. Representa un conjunto de anomalías del desarrollo caracterizado por embriotoxon posterior, hipoplasia del iris, disgenesia de la cámara anterior y glaucoma en un 50 % de los casos.
La hipoplasia del iris puede variar desde un adelgazamiento del estroma medio a una marcada atrofia con formación de agujeros pseudopolicoria, corectopia, ectropión uveal, microcórnea y megalocórnea (Fig. 2). El ángulo camerular es abierto, sin embargo puede haber una inserción alta del iris. Otros hallazgos oculares menos frecuentes son estrabismo, catarata, quistes dermoides, desprendimientos de retina, degeneraciones maculares, colobomas corioretinales e hipoplasias de la cabeza del nervio óptico.
Los hallazgos no oculares son actualmente más consistentes y deben ser siempre buscados en pacientes con estas anomalías oculares. Entre los mismos se encuentran los siguientes:
]]> Existen numerosos síndromes asociados a anomalías de Axenfeld-Rieger como son la distrofia miotónica, el defecto septal atrial y la pérdida auditiva neurosensorial.2,4,9 Esta condición puede ocurrir esporádicamente, aunque el patrón de herencia más frecuentemente es autosómico dominante con alta penetrancia y amplia expresividad.- Anomalías dentarias y faciales: microdontias, anodontias e hipodontias, hipoplasia del maxilar, hipertelorismo, frente prominente, telecantus, nariz ancha y plana (Fig. 2 B y C).
- Anomalías umbilicales: falla en la involución de la piel periumbilical, en algunos se encuentra una piel redundante periumbilical y en otros, en menor cuantía, hernias umbilicales.
- Hipospadias y estenosis anal: estos hallazgos se han presentado con alguna frecuencia.
- Disminución de la hormona del crecimiento: corta estatura y quistes aracnoideos paraselares.
Tres locus cromosómicos han sido implicados en la génesis del SAR: 4q25 (RIEG1), 6p25 y 13q14 (RIEG2). Los genes implicados en los cromosomas 4q25 y 6p25 han sido identificados como PITX2 y FKHL7 (también conocido como el gen FOXC1) respectivamente. El sitio más común de alteración cromosómica ha sido el brazo largo del cromosoma 4, y la traslocación la alteración más común. Algunas deleciones en el cromosoma 13q se han asociado con el SAR, pero hasta el momento no se ha aislado algún gen específico en ésta región.1,6,11-15
Recientemente se ha identificado un cuarto locus en el cromosoma 11, que codifica para un factor de transcripción, el PAX6. Pequeñas deleciones en este gen se relacionan con la presentación del SAR. Con respecto a las alteraciones dentarias se ha implicado el gen productor del factor de crecimiento epidérmico y el gen PITX2 como agentes causales.9,15,16
El tratamiento dependerá de las complicaciones presentadas: pupiloplastia, tratamiento de la ambliopía, tratamiento del glaucoma que es muy difícil en estos casos. Las goniotomías resultan ineficaces, se prefieren medicamentos supresores del humor acuoso y valorar cirugía filtrante o implantes valvulares.
Ectropión congénito del iris.
Es una condición rara, usualmente unilateral, en la que hay un ectropion congénito no progresivo del epitelio pigmentario posterior del iris sobre su superficie anterior.2,12,13,17 Ocurre debido a una hiperplasia no traccional del epitelio pigmentario posterior del iris. Es circunferencial y en ocasiones da la impresión de una anisocoria por su color oscuro.
Se puede acompañar de hipoplasia del iris, inserción alta del iris y glaucoma.18 La pupila del ojo afectado responde a la luz y a la acomodación, aunque en menor intensidad que el ojo sano. Las asociaciones sistémicas que deben ser excluidas son la neurofibromatosis tipo I y el síndrome de Prader Willi.
No tiene un origen genético definido. El manejo del glaucoma es también muy difícil, la terapia médica es insatisfactoria y la técnica quirúrgica que se prefiere es la trabeculectomía con antimetabólitos.
Distrofia endotelial hereditaria congénita.
La distrofia endotelial hereditaria congénita (DEHC) constituye la ausencia completa o casi completa del endotelio corneal. Aparece con edema corneal bilateral difuso que no resuelve o mejora en presencia de presión intraocular normal. Se clasifica en dos tipos: 2,12,15,19,20
]]>- DEHC tipo 1: aparece un tiempo después del nacimiento y tiene relativamente buen pronóstico. Autosómica dominante.
- DEHC tipo 2: comienzo temprano con gran afectación de la visión y nistagmo. Autosómica recesiva.
Varios autores han reportado mutaciones a nivel del cromosoma 20 (20p11.2-20q11.2 y 20p.13).2 El tratamiento quirúrgico, queratoplastia penetrante, será en casos extremos con afectación visual, por los pocos resultados favorables de este proceder en niños pequeños.
Distrofia polimorfa posterior.
Es una enfermedad hereditaria, autosómica dominante y bilateral del endotelio corneal y la membrana de Descemet.2,13,15,21 Los niños pequeños pueden presentar pérdida de la visión por turbidez corneal. Se descubre muchas veces de forma incidental en un adulto asintomático. Esta cursa con aumento de la presión intraocular en aproximadamente el 15 % de estos pacientes.19
Aún no se ha determinado el gen causante pero se ha encontrado un defecto en el cromosoma 20q11. La mayoría no requiere tratamiento. Cuando presentan edema corneal severo se indica queratoplastia penetrante.
Glaucoma congénito primario.
Consiste en un aumento de la presión intraocular, donde encontramos un desarrollo anómalo de las estructuras del ángulo de la cámara anterior. Esto trae como consecuencia alteraciones morfológicas del globo ocular y afectación del nervio óptico. Se caracteriza por la tríada de lagrimeo, fotofobia y blefarospasmo.22
Presenta una incidencia de 1:10 000 nacidos vivos. Se han identificado tres locus, GLC3A(2p21), GLC3B(1p36) y más recientemente GLC3C(14q24).23-26 Stoilov y otros han descubierto mutaciones en el gen CYP1B1 y en el GLC3A de familias conectadas.24,26,27 El tratamiento es quirúrgico con variantes que van de la goniotomía a los implantes valvulares.
]]> Síndromes endoteliales iridocorneales.Tienen en común que son unilaterales, más frecuentes en mujeres y casi exclusivos de la raza blanca. Sin embargo, la microscopia especular revela anomalías corneales e iridianas leves en el ojo "sano". Se reportan mutaciones en los genes SCN1A, BIRC3, IL1B, CASP12.28 El tratamiento del glaucoma será con cirugías filtrante y antimetabólitos, o implantes valvulares preferiblemente.
Representa un conjunto de enfermedades que presentan una anomalía primaria de la córnea y son las siguientes:7,15
- Atrofia progresiva de iris: presenta atrofia del iris progresiva, corectopia, pseudopolicoria y ectropión de iris. La córnea puede aparecer normal o con leve daño endotelial similar a la distrofia de Fuchs.
- Endotelización de las estructuras del segmento anterior con sinequias periféricas anteriores. El glaucoma no es infrecuente y su severidad estará en relación con la extensión de las sinequias.19
- Síndrome de Chandler: asociado con edema corneal debido a los daños corneales endoteliales, cambios del iris leves y glaucoma que responde mejor a la terapia medicamentosa.
- Síndrome del nevus de iris: cambios en el iris de tipo nodular a tipo pigmentado aplanado. Esto puede estar asociado con grados variables de atrofia del iris y cambios del endotelio corneal.
ANOMALÍAS DEL SEGMENTO ANTERIOR DE ORIGEN ECTODÉRMICO
]]> Quistes dermoides limbales y corneales.Los dermoides contienen elementos del ectodermo como epitelio queratinizado, pelos, glándulas sebáceas y sudoríparas, nervios, músculos y rara vez tejido dentario. También además de tejido de origen mesodérmico, como tejido fibroso, grasa, vasos sanguíneos y cartílago.
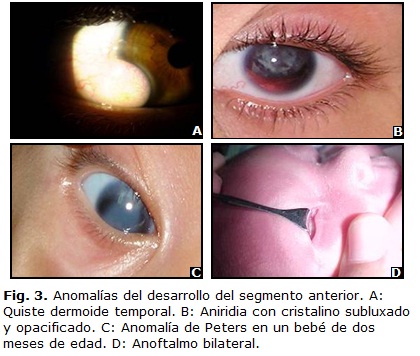
El aspecto clínico es de una masa redonda u ovoide, blanca amarillenta, que puede localizarse en cualquier parte de la superficie ocular. (Fig. 3 A)4,7,22 Tienden a situarse en el limbo, llegan inclusive a rodearlo, aunque su localización más frecuente es en el lado inferotemporal. El segundo sitio por frecuencia es el margen superotemporal de la órbita.
Los dermoides no muestran crecimiento o muy poco. Sin embargo, el aumento de tamaño suele darse en la pubertad. No sufren transformación maligna. Estos se asocian al síndrome de Goldenhar. El tratamiento es quirúrgico.
ANOMALÍAS DEL DESARROLLO DEL SEGMENTO ANTERIOR DE ORIGEN GLOBAL
Megalocórnea congénita.
Puede aparecer aislada o asociada a anomalías del segmento anterior. Cuando se muestra aislada consiste en un alargamiento de la córnea no progresivo, con un diámetro corneal mayor de 12 mm en el recién nacido o mayor de 13 mm a cualquier edad, sin aumento de la presión intraocular, ni rupturas de la membrana de Descemet.7
La miopía es el defecto refractivo más común asociado a esta condición, es común el astigmatismo a favor de la regla. Otros hallazgos oculares incluyen pigmentación a nivel de la malla trabecular, glaucoma de comienzo tardío, hipoplasia del estroma del iris, catarata, ectopia lentis, entre otros.2,7,22 Entre las asociaciones sistémicas están el síndrome de Alport, síndrome de Marfan, síndrome de Down, síndrome de retardo mental y megalocórnea, mucolipidosis tipo II y craneosinostosis.
]]> La megalocórnea congénita ha sido categorizada en cuatro tipos:2- Tipo 1: síndrome de Neuhauser asociado con megalocórnea, hipoplasia de iris y retardo mental.
- Tipo 2: megalocórnea asociada con escoliosis y retardo del crecimiento.
- Tipo 3: megalocórnea asociada con hipotonía severa y microcefalia.
- Tipo 4: megalocórnea asociada con megaloencéfalo y obesidad.
Se reporta asociación entre las regiones DXS87 y DXS94 en los cromosomas Xq21.3-q22 y la megalocórnea.2 El manejo consiste en la observación y seguimiento para detectar complicaciones. Si se refiere fotofobia, indicar lentes oscuros y corregir el defecto refractivo.
Microcórnea.
Es una condición infrecuente y se caracteriza por un diámetro corneal horizontal menor de 10 mm. Puede ser unilateral o bilateral y usualmente se asocia con hipermetropía.2,10
Los hallazgos oculares pueden ser catarata, corectopia, microfaquia, colobomas de iris, retinopatía de la prematuridad, vasculatura fetal persistente y glaucoma. Las asociaciones sistémicas incluyen síndrome de Ehlers-Danlos, síndrome de Marfan, Turner, Rieger, Norrie, Waardenburg, Weill-Marchessani. Se debe corregir el defecto refractivo y la ambliopía en casos aislados. El tratamiento dependerá de las complicaciones presentadas.
]]> Aniridia.Tiene una prevalencia de 1 en 50 000 personas. Se han identificado mutaciones en Pax6 y es el responsable de la aniridia humana (Fig. 3 B). El locus afectado es el 11p13.2,29,30
Es un trastorno bilateral, con ausencia de todo o casi todo el tejido iridiano, aunque puede asociarse a hipoplasia del iris. Se puede presentar hipoplasia del nervio óptico y de la fóvea y resulta en un nistagmo sensorial congénito con afectación de la agudeza visual.2,4,31 Otros hallazgos oculares asociados son catarata polar anterior a menudo con membrana pupilar persistente, catarata cortical, glaucoma, opacidades corneales, subluxación del lente, anomalía de Peter, microcórnea y ectopia lentis.31,32
Puede ser esporádica o familiar. Las dos terceras partes de los niños con aniridia tienen padres afectados. La aniridia esporádica es asociada con tumor de Wilms (asociación de WAGR) en más de un tercio de los casos. Puede encontrarse además con anomalías genitourinarias y retardo mental (síndrome de Gillespie).
El tratamiento es similar a las otras disgenesias, corrección del defecto refractivo con lentes oscuros para reducir la fotofobia y tratamiento del glaucoma que va desde la goniotomía hasta los implantes valvulares. Para la opacificación corneal se recomienda el trasplante de células límbicas o el trasplante corneal.31,32
Queratitis autosómica dominante.
Esta queratitis y la vascularización corneal se deben a mutaciones en PAX6. El niño se presenta con fotofobia, ojo rojo e irritado, se observa una banda corneal circunferencial opaca de 1 a 2 mm y de vascularización corneal contigua con el limbo.
Es de manejo difícil. El examen bajo anestesia se hace necesario para concluir el diagnóstico. Se indican lubricantes oculares y trasplante de células límbicas.7
Córnea plana.
Aplanamiento unilateral o bilateral de la córnea con una curvatura menor de 43 dioptrías. La córnea puede ser clara, o asociada con esclerocórnea y esto afectaría la agudeza visual. Existe una forma dominante y otra recesiva.
]]> Los hallazgos oculares incluyen aniridia, glaucoma, catarata, ectopia lentis, coloboma de iris y coroides, aplasia retinal, sinequia anterior, esclera azul, pseudoptosis y microftalmo. Se asocia a hipermetropía, microcórnea y esclerocórnea.7,14,19 Las asociaciones sistémicas incluyen osteogénesis imperfecta, epidermólisis bulosa, trisomía 13 y síndrome de Hurler.Como se mencionó anteriormente no todo está dicho en la genética ocular. Hay estudios negando la asociación de córnea plana con las mutaciones en DCN, DSPG3, FOXC1, KERA, LUM y PITX2, y otros relacionando mutaciones en KERA en córnea plana autosómica recesiva.33 La conducta a seguir consiste en refracción ciclopléjica, corrección del defecto refractivo, vigilancia del glaucoma y queratoplastia penetrante por la esclerocórnea si es necesario.
Esclerocórnea.
Anomalía infrecuente, no inflamatoria ni progresiva. En esta hay extensión del tejido escleral opaco epiescleral y conjuntival a la periferia de la córnea oscureciendo el limbo. Es bilateral en el 90 % de los casos. La agudeza visual se afecta si el centro de la córnea esta tomado. Puede ser autosómica dominante o recesiva con 50 % de casos esporádicos.2,6,14
Se divide en tres tipos:
- Tipo I: periférica y asociada con córnea plana.
- Tipo II: periférica o central con desorganización y microftalmo.
- Tipo III: leve y periférica solamente.
Puede aparecer aislada o asociada a anomalías oculares y sistémicas. Asociaciones oculares incluyen glaucoma, esclera azul, catarata, colobomas de iris y coroides, aniridia, anomalías del ángulo, microftalmo y córnea plana en el 80 % de los casos. Las asociaciones sistémicas no son tan frecuentes e incluyen anomalías craneales, hipoacusia, espina bífida oculta, anomalías pulmonares, en la cara y piel, síndrome de HallermannStreiff, síndrome de Mieten y síndrome de Smith-Lemli-Opitz.
]]> Las causas genéticas no están precisadas. Su manejo consiste en refracción ciclopléjica, corrección del defecto refractivo, vigilancia del glaucoma y queratoplastia penetrante si es necesario.Anomalía de Peters.
Opacidad corneal central congénita causada por la ausencia de estroma corneal posterior, endotelio y Descemet (Fig. 3 C). El 80 % de los casos son bilaterales y el glaucoma se presenta en el 50 a 70 % de los pacientes.
Se clasifica en tres tipos: 6,12,19,20
- Defecto corneal posterior con leucoma corneal central. Forma más simple y menos documentada en la literatura.
- Defecto corneal posterior con leucoma y adherencias del iris.
- Defecto corneal posterior con leucoma, adherencias del iris y contacto queratolenticular o catarata.
Las asociaciones oculares incluyen síndrome de Axenfeld-Rieger, aniridia, microftalmo, vasculatura fetal persistente y displasia retinal. Entre las asociaciones sistémicas se mencionan las anomalías craneofaciales, enfermedad cardiovascular congénita, hipoplasia pulmonar, anomalías genitourinarias y sindactilia.
La anomalía de Peters bilateral es a menudo asociada con un síndrome, se encuentra microftalmía, y lesiones cutáneas lineales rojizas. Síndrome Peters Plus es un desorden autosómico recesivo que incluye anomalía de Peters bilateral, corta estatura, retardo mental, braquidactilia, braquimorfismo y pérdida de la audición.
]]> En la misma familia existen miembros con retardo mental, labio leporino y paladar hendido. En estos pacientes se reportan mutaciones en el gen B3GALTL.34 También se refiere mutación del gen PAX6 en el cromosoma 11p13, gen PITX2 Y CYPIBI. 2 El manejo de estos pacientes es complejo. Los resultados de la queratoplastia dependen de la habilidad para controlar el glaucoma asociado.Microftalmo
El tamaño del ojo se encuentra disminuido en todos sus diámetros. Suele asociarse a microcórnea sin embargo, puede existir microftalmo con córnea normal y microcórnea sin microftalmo.14
Se relaciona con alteraciones en el gen STRA6 (locus 15q24.1).35 Asociado a anomalías sistémicas como acondroplasia, retardo mental, embriopatía diabética, micropolisacaridosis VI y mucolipidosis III.
Anoftalmo.
Parece faltar el globo ocular, pero la disección cuidadosa suele revelar la existencia de una pequeña masa rudimentaria sólida o quística (Fig. 3 D).14 Generalmente bilateral y esporádico. Está relacionado con mutación del gen SOX2.36,37 El tratamiento será con fines estéticos mediante la colocación de prótesis oculares.
CONCLUSIÓN
En todas las anomalías del desarrollo del segmento anterior tratadas apreciamos que además de la clínica, es cada vez más importante y posible debido a los adelantos de la genética molecular la identificación de genes. Esto ha hecho posible la clasificación de estas disgenesias según la mutación genética subyacente. Debido a la variabilidad de cuadros clínicos, estos estudios ayudan a confirmar el diagnóstico de la disgenesia que presenta el paciente y así permiten brindarle un tratamiento más adecuado.
]]> REFERENCIAS BIBLIOGRÁFICAS
1. Alward WL. Axenfeld-Rieger Syndrome in the age of molecular genetics. Am J Ophthalmol. 2000 Jul [citado 30 may 2009];130(1). Disponible en: http://www.ajo.com/article/S0002-9394(00)00525-0/abstract
2. Idrees F, Vaideanu D, Fraser SG, Sowden JC, Khaw PT. A Review of anterior segment dysgeneses. Survey of Ophthalmology. 2006 May-Jun [citado 30 may 2009];51(3). Disponible en: http://www.sciencedirect.com/science/article/pii/S0039625706000324
3. Rapuano CJ, Luchs JL, Kim T. Los requisitos en segmento anterior. Madrid: Harcourt; 2001.
4. Nelson LB. Enfermedades de la Córnea. En: Nelson LB. Harley, Oftalmología Pediátrica. 4ta ed. Philadelphia: Mc Graw Hill Int; 2000. p. 242-90.
5. American Academy of Ophthalmology. Pediatric Ophthalmology and Strabismus. USA: American Academy of Ophthalmology; 2008 (Basic and clinical Science Course).
6. Elhilaly H, Essawy R. Congenital Anomalies of the anterior segment. En: Garg A. Surgical and medical Management of pediatric Ophthalmology. New Delhi: Jaypee; 2007. p. 409-26.
7. Babak E. Mesenchymal Dysgeneses. En: Garg A. Surgical and medical Management of pediatric Ophthalmology. New Delhi: Jaypee; 2007. p. 1214-24.
8. Genetics Home Reference [Internet]. Iridodigenesis [actualizado 2010; citado 18 dic 2010]. Disponible en http://ghr.nlm.nih.gov/search?query=iridogoniodysgenesis
9. Nischal K, Sowden J. Anterior Segment: Develpmental Anomalies. En: Pediatric Opthalmology and Strabismus. Philadelphia: Elsevier Ltd; 2005. p. 249-64.
10. Ortiz GE, Milena G. Síndrome de Axenfeld-Rieger con glaucoma bilateral y descompensación de córnea en ojo izquierdo. Rev Fac Med Univ Nac Colomb. 2004 [Citado 10 septiembre 2009];52(3). Disponible en: http://www.imbiomed.com/1/1/articulos.php?method=showDetail&id_articulo=29523&id_seccion=1981&id_ejemplar=3027&id_revista=121
11. Eguía F, Rio M, Capote A. Manual de Diagnóstico y Tratamiento en Oftalmología. La Habana: ECIMED; 2009.
12. Schargel K, Hueso JR. Malformaciones Oculares y disgenesias de cámara anterior. En: Fonseca Santodomingo A, Abelairas Gómez J, Rodríguez Sánchez J, Peralta Calvo J. Actualización en Oftalmología Pediátrica. España:Tecnimedia Editorial, S.L; 2000. p.33-51.
13. Garcia-Rubio L, Gracia E, Tejada P. Patología corneal en niños. En: Fonseca Santodomingo A, Abelairas Gómez J, Rodríguez Sánchez J, Peralta Calvo J. Oftalmología Pediátrica. España:Tecnimedia Editorial, S.L. 2000 . p.281-6.
14. Espinoza HM, Cox CJ, Semina EV, Amendi BA. A molecular basis for differential developmental anomalies in AxenfeldRieger Syndrome. Hum Mol Genet. 2002 Apr [Citado 10 sep 2009];11(7). Disponible en: http://hmg.oxfordjournals.org/content/11/7 /743.full.pdf+html
15. Lines MA, Kozlowski K, Walter MA. Molecular genetics of Axenfeld-Rieger malformations. Hum Mol Genet. 2002 May [citado 30 agosto 2009]15;11(10). Disponible en: http://hmg.oxfordjournals.org/content/11/10/1177.full.pdf+html
16. Lines MA, Kozlowski K, Kulak SC, Allingham RR, Heon E, Ritch R, et al. Characterization and prevalence of PITX2 microdeletions and mutations in Axenfeld-Rieger malformations. Invest Ophthalmol Vis Sci. 2004 [citado 30 ago 2009];45(3). Disponible en: http://www.iovs.org/content/45/3/828.short
17. Wilson ME. Congenital iris ectropion and a new classification for anterior segment dysgeneses. J Pediatr Ophthalmol Strabismus. 1990 [Citado 30 agosto 2009];27(1). Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/2324918
18. Monaco G, Franceschin S, Cacioppo V, Simonetta S, Ratiglia R. Congenital iris ectropion associated with juvenile glaucoma. J Pediatr Ophthalmol Strabismus. 2009 Jan-Feb [Citado 30 agosto2009];46(1). Disponible en http://www.ncbi.nlm.nih.gov/pubmed/19213275
19. Wright K, Spiegel P. Enfermedad de la córnea y el segmento anterior. En: Wright K, Spiegel P. Los requisitos en Oftalmología pediátrica y estrabismo. ST. Louis: Mosby; 2001. p. 47-59.
]]>20. Meyer I, Rolim H, Medeiros A, Paiva L, Galvão FR. Peter's anomaly, clinical and therapeutic aspects: case report. Arq Bras Oftalmol. 2010 [citado 29 sep 2009];73(4). Disponible en http://www.scielo.br/scielo.php?script=sci_issuetoc&pid=0004-274920100004&lng=en&nrm=iso
21. Díaz JL, Turati M, Turati M. Opacidad corneal en niños. Informe de un caso de distrofia endotelial congénita. Acta Pediatr Mex. 2006 [citado 29 sep 2009];27(3):125-7.
22. Kanski JJ. Oftalmología clínica. 5ta. ed. Madrid: Elsevier; 2004.
23. González-Huerta LM, Messina-Baas OM, Lara Huertas SF, Babayan Mena JI, Cuevas-Covarrubias SA. Glaucoma Congénito Primario. Rev Mex Oftalmol. 2005 [citado 29 sep 2009];79(2):106-10. Disponible en http://www.imbiomed.com.mx/1/1/articulos.php?method=showDetail&id_articulo=31823&id_seccion=851&id_ejemplar=3272&id_revista=31
24. Ben-Zion I, Bogale A, Moore DB, Helveston EM. Bilateral primary congenital glaucoma in monozygotic twins. J Pediatr Ophthalmol Strabismus. 2010 Mar-Apr [citado 30 ago 2009];47(2). Disponible en http://www.ncbi.nlm.nih.gov/pubmed/20349909
25. Stoilov IR, Costa VP, Vaconcellos JP, Melo MB, Betinjane AJ, Carani JC, et al. Molecular genetics of primary congenital glaucoma in Brazil. Invest Ophthalmol Vis Sci. 2002 [citado 10 dic 2009];43(6). Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/12036985
26. Wiggs JL. Genetics etiologies of Glaucoma. Arch Ophthalmol. 2007 [citado 10 dic 2009]:125(1). Disponible en: http://archopht.ama-assn.org/cgi/content/full/125/1/30
27. Choudhary D, Jansson I, Schenkman JB, Sarfarazi M, Stoilov I. Comparative expression profiling of 40 mouse cytochrome P450 genes in embryonic and adult tissues. Arch Biochem Biophys. 2003 [citado 30 ago 2009];414(1). Disponible en http://www.ncbi.nlm.nih.gov/pubmed/12745259
28. González Y. Glaucoma Pediátrico. Bases Genéticas. En: Río Torres M. Oftalmología. Criterios y Tendencias Actuales. La Habana: ECIMED; 2009. p. 671-8.
29. Genetics Home Reference [Internet]. Síndrome Iridocorneoendotelial [actualizado 2010; citado 18 dic de 2010]. Disponible en http://ghr.nlm.nih.gov/search?query=ICE
30. Kokotas H, Petersen MB. Clinical and molecular aspects of aniridia. Clinical Genetics. 2010 [citado 10 dic 2009];77(5). Disponible en http://onlinelibrary.wiley.com/doi/10.1111/j.1399-0004.2010.01372.x/abstract
31. Brauner SC, Walton DS, Chen TC. Aniridia. International Ophthalmology Clinics. 2008 [citado 19 ene 2011];48(2). Disponible en http://journals.lww.com/internat-ophthalmology/Citation/2008/04820/Aniridia.11.aspx
32. Lee H, Meyers K, Lanigan B, O'Keefe M. Complications and visual prognosis in children with aniridia. Journal of Pediatric Ophthalmology and Strabismus. 2010 Jul-Aug [citado 19 ene 2011];47(4). Disponible en http://www.slackjournals.com/article.aspx?rid=43065
33. Lee H, Khan R, O'Keefe M. Aniridia: current pathology and management. Acta Ophthalmológica. 2008 [citado 19 ene 2011];86(7). Disponible en: http://onlinelibrary.wiley.com/doi/10.1111/j.1755-3768.2008.01427.x/pdf
34. Aldave AJ, Sonmez B, Bourla N, Schultz G, Papp JC, Salem AK, et al. Autosomal dominant cornea plana is not associated with pathogenic mutations in DCN, DSPG3, FOXC1, KERA, LUM, or PITX2. Ophthalmic Genetics. 2007 [citado 19 ene 2011];28(2). Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/17558846
35. Genetics Home Reference [Internet]. Peters plus syndrome [actualizado 2010; citado 18 dic 2010]. Disponible en: http://ghr.nlm.nih.gov/condition/peters-plus-syndrome
36. Genetics Home Reference [Internet]. STRA6; [actualizado 2010; citado 18 dic de 2010]. Disponible en: http://ghr.nlm.nih.gov/gene/STRA6
37. Genetics Home Reference [Internet]. SOX2 anophthalmia syndrome; [citado 18 dic 2010]. Disponible en: http://ghr.nlm.nih.gov/condition/sox2-anophthalmia-syndrome
Recibido: 22 de febrero de 2011.
Aprobado: 13 de septiembre de 2011.
Dra. Lourdes R Hernández Santos. Instituto Cubano de Oftalmología "Ramón Pando Ferrer". Ave. 76 No. 3104 entre 31 y 41 Marianao, La Habana, Cuba. Correo electrónico: lourdesrita@infomed.sld.cu
]]>