Enfermedad de Rendu Osler Weber: presentación de un caso
Rendu Osler Weber disease: one case report
Dra. Lidia Duque Estrada; Dr. Nguyen Castro Gutiérrez; Dr. José Larquin Comet; Dr. Miguel Damián Junco Bonet; Dr. Gilberto Betancourt Reyes
Hospital Provincial Universitario Manuel Ascunce Domench. Camagüey, Cuba.
]]>
RESUMEN
Fundamento: la telangiectasia hemorrágica hereditaria o enfermedad de Rendu-Osler-Weber es una enfermedad autosómica dominante caracterizada por la presencia de múltiples telangiectasias en piel y mucosas, asociadas a malformaciones arteriovenosas de distintos órganos, incluidos pulmones, sistema gastrointestinal y sistema nervioso central. Su pronóstico es incierto, con un diagnóstico y tratamiento precoz es posible mejorar la calidad de vida del paciente y lograr una expectativa de vida similar a la de la población general.
Objetivo: presentar un caso con el diagnóstico de telangiectasia hemorrágica hereditaria.
Caso Clínico: paciente femenina de 64 años de edad con diagnóstico de telangiectasia hemorrágica hace 17 años, con episodios de epistaxis frecuente, antecedentes familiares de la enfermedad (padre, tíos paternos y hermanos), que ingresa en el servicio de cuidados intermedios con cuadro de epistaxis y manifestaciones clínicas de anemia aguda, lesiones nodulares en manos, dedos y pabellón auricular, con telangiectasias en la lengua.
Conclusiones: la telangiectasia hemorrágica hereditaria es una enfermedad poco frecuente pero existen reportes de casos a nivel mundial.
DeCS: TELANGIECTASIA HEMORRÁGICA HEREDITARIA/diagnóstico; MALFORMACIONES ARTERIOVENOSAS; EPISTAXIS; ABERRACIONES CROMOSÓMICAS; ANCIANO.
ABSTRACT
Background: hereditary haemorrhagic telangiectasia (HHT) or Rendu Osler Weber Sickness is a dominant autosomic illness characterized by the presence of multiple telangiectasias on skin and mucus, associated to arterovenous malformations in different organs, including lungs, central nervous system and gastrointestinal system. Its prognosis is uncertain. It is possible to improve the quality of life by diagnosing and treating it early, therefore a life expectancy similar to the general population can be reached.
Objetive: to present a case with a diagnosis of HHT. ]]>
Clinical case: A sixty-four-year-old female patient with a diagnosis of HHT 17 years ago, with episodes of frequent epistaxis and a family history of HHT (father, uncles on the father´s side and brothers) is admitted to the ICU with clinical manifestations, acute anemia and nodule lesions on her hands, fingers and auricular pavillion, with telangiectasias on the tongue.
Conclusion: HHT is not a common illness but there are cases reported all over the world.
DeCS: TELANGIECTASIA, HEREDITARY HEMORRHAGIC/diagnosis; ARTERIOVENOUS MALFORMATIONS; EPISTAXIS; CHROMOSOME ABERRATIONS; AGED.
INTRODUCCIÓN
La telangiectasia hemorrágica hereditaria (THH) o enfermedad de Rendu-Osler-Weber fue descrita por primera vez en el año 1876, por John Wickham Legg y después, por Henri Jules Renduen 1896 citada por Shovlin C. 1 En el año 1901, William Osler citado por Geisthoff UW, et al, 2 describió tres pacientes que padecían una forma familiar rara de epistaxis (hemorragias nasales) recurrente, asociada con telangiectasias (dilatación de los vasos sanguíneos de tamaño muy pequeño que daba lugar a manchas de color púrpura con aspecto de araña) en piel y mucosas.
Se trata de una angiopatía neoformativa de telangiectasias circunscritas que al romperse, determinan síndromes hemorrágicos locales. La THH es infrecuente, se estima una prevalencia de dos casos por 100 000 personas, es mayor en algunas áreas geográficas, como en la isla danesa de Fyn, las Antillas Danesas y en algunas regiones de Francia. 3
Esta enfermedad afecta de forma primordial a pacientes caucásicos, aunque existen reportes ocasionales en pacientes asiáticos y árabes. Afecta por igual a ambos sexos, comienza con más frecuencia durante la pubertad o adultez, entre los 20 y los 40 años, aunque también puede presentarse en niños. 3, 4
La THH es una enfermedad vascular rara, congénita, que se caracteriza por la presencia de múltiples telangiectasias con tendencia a sufrir hemorragias localizadas en la nariz y en las vías urinarias, aunque también es posible que se desarrollen anomalías vasculares internas al nivel del cerebro, pulmones, garganta, laringe, tracto gastrointestinal, hígado, vejiga y vagina. Las telangiectasias suelen verse en labios, lengua y mucosa nasal, y también pueden afectar otras zonas como la cara y las orejas. 5
]]> De forma genética presenta un rasgo autosómico dominante, por lo que basta que uno de los padres la padezca para que el hijo pueda resultar enfermo. 6 Se trata de un gen individual, anormal en uno de los cromosomas autosómico (uno de los primeros 22 cromosomas no sexuales). En su patogénesis están implicados dos genes, THH1 y THH2, los cuales determinan dos formas diferentes de una misma enfermedad. La variante THH1 se origina por mutaciones en el gen endoglina (ENG), localizado en el brazo largo del cromosoma nueve (9q33-q34.1), mientras que el THH2 es causado por mutaciones en el gen ALK1, localizado en el brazo largo del cromosoma 12 (12q11-q14). 6, 7Se caracteriza, desde el punto de vista clínico, por la presencia desde el nacimiento, de múltiples telangiectasias en piel y mucosas (dilataciones venulares y capilares), propensión a hemorragias localizadas, de manera especial en fosas nasales, urinarias (hematuria) y con menor frecuencia, digestivas (gastrorragia) y respiratorias (hemoptisis). 8
El objetivo de esta investigación es presentar un caso con el diagnóstico de telangiectasia hemorrágica hereditaria.
CASO CLÍNICO
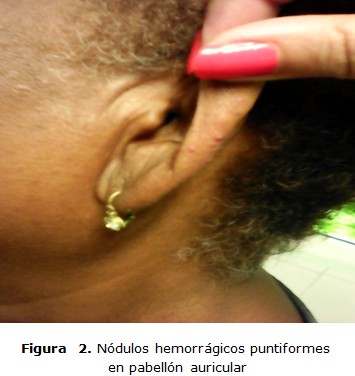
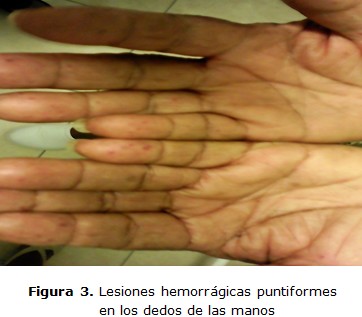
Paciente femenina de 64 años de edad, de raza negra con antecedentes patológicos personales de THH hace 17 años, con episodios de epistaxis frecuente entre tres o cuatro al año con antecedentes familiares de la enfermedad (padre, tíos paternos y hermanos); ingresa en el servicio de cuidados intermedios con cuadro de sangramiento nasal, unilateral derecho y gingival, acompañado de astenia marcada, debilidad y fatiga. Examen físico: se constata sangrado activo de moderada intensidad, por fosa nasal derecha y mucosa gingival con palidez cutánea mucosa severa, lesiones nodulares puntiformes no dolorosas y blandas en borde cubital de las manos y pulpejo de los dedos (figura 3), así como en el pabellón auricular (figura 2) y una lesión de aspecto telangiectásico, papular y anular de 1 cm de diámetro en la cara lateral de la lengua (figura 1), taquicardia sinusal con frecuencia cardíaca de 110 lpm, tensión arterial 110/70 mmHg.
Estudios analíticos ]]> Hematocrito: 0,20 %, plaquetas: 433 x109/L, leucocitos: 12 x 109/l anisopoiquilocitosis, hipocromía marcada, eritrosedimentación: 65 mm/h, ácido úrico: 335 mmol/l, colesterol: 5,6 mmol/l triglicéridos: 1,35 mmol/l, amilasa pancreática: 78 u/l, transaminasa glutámico pirúvica: 16 u/l, transaminasa glutámico oxalacética: 18 u/l, fosfatasa alcalina: 120 u, glucosa: 5,6 mmol/l, albumina: 35g/l, coagulograma: normal, serología: no reactivo, hierro sérico: 3,8 mmol/l.
Estudios imaginológicos
Rayos X de tórax: no alteraciones pleuropulmonares ni cardiovasculares, panendoscopia: no lesiones de telangiectasia.
Se diagnostica por clínica una anemia aguda secundaria a epistaxis y gingivorragia, con hierro sérico bajo en el curso de una THH. Se comienza la terapéutica con medidas generales reposición de glóbulos rojos, tratamiento con hierro parenteral, taponamiento anterior en fosa nasal derecha por 48 horas y eritropoyetina recombinante por tratarse de un sangramiento crónico con agudización frecuente.
DISCUSIÓN
Esta enfermedad afecta a pacientes caucásicos, aunque existen reportes publicados por Amieva M, et al. 3 Aunque se ha descrito en pacientes asiáticos y árabes. 4 La condición homocigota es muy probable que sea letal. 9 De manera eventual no hay historia familiar de THH, lo que podría ser explicado por una mutación espontánea, historia familiar incompleta o penetrancia incompleta de la enfermedad. 10 Geisthoff U, et al, 11 en estudios recientes sobre el manejo de la THH, se han identificado mutaciones en, al menos, dos genes en diferentes familias con la enfermedad. Un primer grupo tiene mutado el gen de la endoglina (ENG), codificado en el cromosoma 9, donde se presenta con mayor frecuencia malformaciones arteriovenosas (MAV) pulmonares; un segundo grupo tiene mutado el gen que codifica el receptor de activina A ubicado en el cromosoma 12, donde se expresa un fenotipo más leve y un inicio más tardío de la enfermedad. 10, 11 Ambos genes codifican una glicoproteína integral de membrana que se expresa en células del endotelio vascular y que actúa como un receptor de superficie para el factor de crecimiento transformante β (TGF-β). 10 La función de ambas proteínas y la señalización por el TGF-β son esenciales para una angiogénesis normal. Sin embargo, no están disponibles para la práctica clínica, técnicas para el diagnóstico molecular de la enfermedad, 12 la característica clínica más importante es el sangrado de las mucosas, el que es recurrente, espontáneo o producido por un traumatismo menor. Puede comenzar a cualquier edad, donde es más común en la tercera década de la vida. 13 La epistaxis es la forma de sangrado más frecuente (80 %), seguida de hemorragias gastrointestinales (10 a 40 %), genitourinarias, pulmonares e intracerebrales, el compromiso hepático en pacientes con THH resulta de manera primordial de cortocircuitos entre la arteria hepática y las venas hepáticas. (Menos del 10 % cada una). 14
La hemorragia intraocular es rara. Alrededor de un 10 % de los pacientes afectados nunca reporta un sangrado. 15 La piel y las superficies mucosas presentan múltiples telangiectasias hasta en el 89 % de los pacientes. Las telangiectasias pueden ser aracneiformes, lineales o puntiformes y de manera frecuentes son subdiagnosticadas, estas son más frecuentes en la cara, labios, lengua, lecho ungueal, dedos y mucosa nasal. 16 Las lesiones cutáneas por lo general aparecen en la tercera década de la vida, aumentan en tamaño y cantidad con la edad y sangran con facilidad. 17 Entre el 50 y 90 % de los pacientes con THH presenta epistaxis recurrente, donde muchas veces la forma de presentación de la enfermedad, es por lo general antes de los 10 años de edad. 9, 14
En la mitad de los pacientes la frecuencia y gravedad de la epistaxis aumenta con la edad. Alrededor de un tercio de los pacientes tiene sangrados leves, un tercio tiene sangrados moderados que requieren asistencia médica y el otro tercio presenta hemorragias incoercibles, que requieren hospitalización y transfusiones sanguíneas. 18 La epistaxis proviene de pequeñas telangiectasias nasales, las cuales pueden desarrollarse antes que las lesiones cutáneas sean detectables y a menudo aparecen un año antes del primer episodio de epistaxis. 19 La enfermedad de Rendu-Osler-Weber es el trastorno que con frecuencia se asocia a fístulas arteriovenosas pulmonares en niños y adultos. Entre el 15 y 33 % de los pacientes con THH presenta MAV pulmonares, el 70 % de éstas se localizan en bases pulmonares y pueden producir un cortocircuito de derecha a izquierda importante, con hipoxemia significativa, que se puede manifestar por disnea, disminución de la capacidad de ejercicio y cianosis. 16, 20 En el examen físico pueden presentar un soplo de baja intensidad a nivel del foco pulmonar al final de la inspiración e hipocratismo digital.
Otras complicaciones incluyen hemoptisis o hemotórax mortales y embolia paradojal con eventos isquémicos cerebrales. 21 Dado que la severidad y las alteraciones presentes en cada paciente son tan variables, el manejo debe ser individualizado. Una vez que se ha hecho el diagnóstico de THH, se recomienda realizar un despistaje en busca de MAV en distintos parénquimas, de manera esencial en el pulmón y sistema nervioso central. Se recomienda administrar suplementos de hierro en todos los pacientes, para prevenir el déficit secundario a los sangrados.
]]> Puede ser necesario suplementar además con ácido fólico si la médula ósea está siendo estimulada en forma crónica por las pérdidas de sangre y en algunos casos se puede requerir de transfusiones de sangre e inmunización contra el virus hepatitis B según reportan Nollen GJ, et al, 20 en un estudio sobre aneurisma de la arteria pulmonar en pacientes con THH.
CONCLUSIONES
La enfermedad de Rendu Osler Weber o THH es una enfermedad poco frecuente según la bibliografía revisada y en la experiencia de los autores es el primer caso con lesiones hemorrágicas en los dedos y pabellón auricular.
REFERENCIAS BIBLIOGRÁFICAS
1. Shovlin C. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev. 2010 Nov;24(6):203-19.
2. Maune S, Schneider G. Hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber disease) as an example of a rare disease relevant for otorhinolaryngology. Laryngorhinootologie. 2011 Apr;90(4):230-42.
]]>3. Amieva M, López AC, Zamario S. Telangiectasia facial y bicitopenia como sospecha de síndrome de Rendu-Osler-Weber. Dermatología Rev Mex. 2010;54(6):342-5.
4. Begbie ME, Wallace GM, Shovlin CL. Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): a view from the 21st century. Postgrad Med J. 2003;79(927):1824.
5. Grand A. Hereditary hemorrhagic telangiectasia. Can Med Asso J. 2009;180(8):833-835.
6. Kamath N, Bhatia S, Singh H. Hereditary Hemorrhagic Telangiectasia. North Ame J Med Sci. 2015;7(3):125-8.
7. Pierucci P, Lenato GM, Suppressa P, Lastella P, Triggiani V, Valerio R, et al. A long diagnostic delay in patients with Hereditary Haemorrhagic Telangiectasia: a questionnaire-based retrospective study. Orphanet J Rare Dis. 2012 Jun 7;7:33.
]]>8. Govani F, Shovlin C . Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet. 2009;17(7):860-71.
9. Yin L, Reh D, Hoag J, Robinson G. The minimal important difference of the epistaxis severity score in hereditary hemorrhagic telangiectasia. The Laryngoscope. a clinical and scientific review. Eur J Hum Genet. 2015;171(4):443-52.
10. Dittus C, Streiff M, Ansell J. Bleeding and clotting in hereditary hemorrhagic telangiectasia. Wor J Clin Cas. 2015;3(4):330-7.
11. Geisthoff U, Nguyen H, Röth A. How to manage patients with hereditary haemorrhagic telangiectasia. Br J Haematol. 2015;171(4):443-52.
12. Stapf C, Mohr J. Unruptured brain arteriovenous malformations should be treated conservativelyyes. Stroke. 2007;38(12):3308-9.
]]>13. Neetika G, Khunger M, Gupta A. Optimal management of hereditary hemorrhagictelangiectasia. J Blood Med. 2014;5(1):191-206.
14. Buonamico P, Suppressa P, Lenato G. Liver involvement in a large cohort of patients with hereditary hemorrhagic telangiectasia: echo-color- Doppler vs multislice computed tomography study. J Hepatol. 2008;8(5):811-20.
15. Wain K, Ellingson M, McDonald J, Gammon A, Roberts M, Pichurin P, et al. Appreciating the broad clinical features of SMAD4 mutation carriers: a multicenter chart review. Genet Med. 2014;16(8):588-93.
16. Livesey J, Manning R, Meek J. Low serum iron levels are associated with elevated plasma levels of coagulation factor VIII and pulmonary emboli/deep venous thromboses in replicate cohorts of patients with hereditary haemorrhagic telangiectasia. Thorax. 2013;67(4):328-33.
17. Hosman A, Devlin H, Silva B. Specific cancer rates may differ in patients with hereditary haemorrhagic telangiectasia compared to controls. Orphanet J Rare Dis. 2013;20(8):195.
]]>18. González Casas R, Trapero Marugán M, Moreno Otero R. Hepatic disease in hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber disease). Med Clin (Barc). 2007;129(16):629-31.
19. Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2014;48(2):73-87.
20. Nollen GJ, Kodde J, Beek AM, JMJ Res. Van Rossu AC. Quadricuspid pulmonary valve and left pulmonary artery aneurysm in an asymptomatic patient assessed by cardiovascular MRI. Neth Heart J 2013;21:196-8.
21. Rao U, Nair SK, Agarwal A. Aneurysm of pulmonary artery. Heart 2013;98(22):1684.
22. Peery W. Clinical Spectrum of hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu disease). Am J Med. 1987;82:989-97.
]]>
Recibido: 20 de octubre de 2016
Aprobado: 17 de noviembre de 2016
Dra. Lidia Duque Estrada. Especialista de I Grado en Medicina Intensiva y Emergencias. Especialista de I grado en Medicina General Integral. Hospital Universitario Manuel Ascunce Domenech. Camagüey, Cuba. Email: nguyen@mad.cmw.sld
]]>