Enfermedad de Gaucher tipo 1. Presentación de caso
Type 1 Gaucher’s Disease. Presentation of a Case
Rodolfo Millán Batista 1, Jenny María Patiño Pérez 2, Martha Julia Matos Pérez 1, Nitza Julia Sanz Pupo 3, Oneida Reyes González 4
1. Especialista de Primer Grado en Anatomía Patológica. Asistente. Hospital Pediátrico Octavio de la Concepción y de La Pedraja. Holguín. Cuba.
2. Especialista de Primer Grado en Anatomía Patológica. Máster en Medios Diagnósticos. Profesora Auxiliar. Hospital General Vladimir Ilich Lenin. Holguín. Cuba.
3. Especialista de Segundo Grado en Anatomía Patológica. Máster en Educación Médica. Máster en Longevidad Satisfactoria. Profesora Auxiliar. Investigadora Agregado. Hospital Vladimir Ilich Lenin. Holguín. Cuba.
4. Especialista de Primer Grado en Anatomía Patológica. Máster en Medios Diagnósticos. Asistente. Hospital General Vladimir Ilich Lenin. Holguín. Cuba.
]]>
RESUMEN
La enfermedad de Gaucher es un trastorno autosómico recesivo raro debido a la ausencia de la enzima glucocerebrosidasa, produciéndose acumulación de glucocerebrósidos en el sistema retículo endotelial. Se manifiesta por alteraciones hemáticas, hepatoesplenomegalia y manifestaciones neurológicas y óseas. Las células que acumulan el glucocerebrósido son llamadas células de Gaucher. Se presenta una paciente femenina, blanca, de 16 años de edad, de procedencia rural que acudió a consulta del Hospital Pediátrico Provincial Octavio de la Concepción y de la Pedraja por presentar anemia, decaimiento y pérdida de peso, acompañado además por aumento de volumen del abdomen. Con el antecedente de presentar necrosis aséptica del fémur derecho, y madre diabética. Al examen físico se constató la presencia de una gran palidez cutánea mucosa, hepatoesplenomegalia. El examen neurológico fue totalmente normal. En los exámenes de sangre se encontró anemia y trombocitopenia. Se realizó biopsia de bazo, hígado y médula ósea encontrándose las células de Gaucher. El diagnóstico se realizó basado en la clínica, cobrando gran peso la presencia de manifestaciones óseas y la hepatoesplenomegalia. Se basó además en la observación histológica de las células de Gaucher y en la determinación de los niveles bajos de actividad de la enzima glucocerebrosidasa en los leucocitos de la sangre o fibroblastos cutáneos.
Palabras clave: Enfermedad de Gaucher, glucocerebrósidos, hepatoesplenomegalia.
ABSTRACT
Gaucher disease is a rare autosomal recessive disorder due to the absence of the enzyme glucocerebrosidase, resulting in accumulation of glucocerebrosides in the endothelial reticulum system. It is manifested by hematic alterations, hepatosplenomegaly and neurological and bone manifestations. Cells that accumulate the glucocerebroside are called Gaucher cells. A 16-year-old white female patient of rural origin who came to Octavio de la Concepción y de La Pedraja Provincial Pediatric Hospital presenting anemia, decay and weight loss, was also accompanied by an increase in volume abdomen. With the history of presenting aseptic necrosis of the right femur and diabetic mother. Physical examination revealed the presence of large mucosal skin paleness, hepatosplenomegaly. The neurological examination was completely normal. Anemia and thrombocytopenia were found on the blood tests. Biopsy of the spleen, liver and bone marrow was performed with Gaucher cells. The diagnosis was made based on the clinic, with heavy presence of bone manifestations and hepatosplenomegaly. It was also based on histological observation of Gaucher cells and on the determination of low levels of glucocerebrosidase enzyme activity in blood leukocytes or cutaneous fibroblasts.
Keywords: Gaucher's disease, glucocerebrosides, hepatosplenomegaly.
INTRODUCCION
]]> La enfermedad de Gaucher es una enfermedad rara. Se refiere a un grupo de trastornos autosómicos recesivos secundarios a mutaciones del locus de la glucocerebrosidasa situado en el cromosoma 1q21. El gen afectado codifica a la glucocerebrosidasa, enzima que separa los residuos de glucosa de la ceramida. La consecuencia del defecto enzimático es una enfermedad de almacenamiento lisosómico, dada por la acumulación de cerebrósido que se produce en las células fagocitarias de todo el organismo, principalmente en las células del sistema retículo endotelial y, en algunas de las variantes de la enfermedad, también en el sistema nervioso central 1.Los glucocerebrósidos son sustancias que se están formando continuamente a partir del catabolismo de los glucolípidos que forman parte de las membranas celulares de leucocitos y hematíes obsoletos. Son un tipo de esfingolípidos, por lo que esta enfermedad se incluye dentro del grupo de las lipidosis que son enfermedades producidas por almacenamiento de lípidos 2-4.
Existen tres tipos de presentación de la enfermedad: 1, 2 y 3 1.
Tipo 1. No neuropática. Es la forma más común y afecta a 1 de cada 40 000 a 60 000 niños nacidos vivos. La enfermedad Tipo I, no afecta el cerebro o al sistema nervioso central, inclusive se ha visto que algunos pacientes con la enfermedad de Gaucher Tipo I carecen de síntomas representativos, mientras que otros desarrollan síntomas severos que pueden ser fatales. Por lo general el cuadro clínico está dominado por la afectación esplénica y esquelética.
Tipo 2. Neuropática aguda. La enfermedad de Gaucher Tipo II es más rara y poco frecuente por eso se presenta en menos de 1 por cada 100 000 niños nacidos vivos (forma cerebral aguda del lactante).
Tipo 3. Neuropática crónica. El Tipo III es también poco común y afecta a menos de 1 por cada 100 000 niños nacidos vivos (intermedia entre las dos anteriores). Suelen ser pacientes jóvenes con la afectación sistémica característica del tipo I y afectación progresiva del SNC característica del tipo II.
Las células que acumulan el glucocerebrósido son llamadas células de Gaucher, localizadas principalmente en médula ósea, hígado, bazo y ganglios linfáticos 1, 5.
Las manifestaciones de la enfermedad de Gaucher incluyen anemia, trombocitopenia, hepatoesplenomegalia y lesiones óseas. Más del 90 % de los pacientes sufren trastornos óseos.1 El malestar, el dolor característico y la discapacidad producidos por estos trastornos óseos afectan gravemente la calidad de vida de quienes la padecen 6-9.
Resulta importante hacer un rápido diagnóstico e instaurar la terapia específica para dicha enfermedad, ya que en ello radica una pronta mejoría de los síntomas y de las deficiencias que presenta el paciente.
El diagnóstico confirmatorio es realizado mediante el ensayo fluorométrico de la actividad de la enzima responsable 10 y la búsqueda de las células de Gaucher. También se realizan estudios genéticos, pero éstos son menos comunes pues se han identificado 200 mutaciones diferentes del gen que codifica la glucocerebrosidasa.
]]>PRESENTACION DE CASO
Paciente femenina, mestiza, de 16 años de edad, de procedencia rural que acude a consulta del Hospital Pediátrico Provincial Octavio de la Concepción y de la Pedraja por presentar anemia, decaimiento y pérdida de peso, acompañado además por aumento de volumen del abdomen. Con el antecedente de presentar necrosis aséptica del fémur derecho o Enfermedad de Legg- Calves- Perthes, para lo cual lleva tratamiento ortopédico específico por 10 años. Tiene antecedente patológico familiar de madre con diabetes mellitus tipo 2.
Al examen físico se constató la presencia de una gran palidez cutánea mucosa así como hepatoesplenomegalia de gran tamaño. El bazo se extendía hasta la región inguinal izquierda, de consistencia dura, firme, no dolorosa. El hígado rebasaba más de 6 cm del reborde costal presentando las mismas características descritas en el bazo, no doloroso, duro y ligeramente nodular. El examen neurológico fue totalmente normal y la paciente se encontraba orientada en tiempo y espacio, con una conducta normal.
En los exámenes complementarios se encontró: Hemoglobina con cifras de entre 50 y luego 70 g/l. Hay evidencia en el momento del ingreso de una trombocitopenia en sangre periférica. Se observó en la ultrasonografía la hepatoesplenomegalia y a los Rx se pudieron apreciar las lesiones de la necrosis aséptica ósea del fémur derecho.
Se le realizó punción con aguja fina del bazo, biopsia de hígado y extendido citológico y biopsia de médula ósea con el fin de conocer el diagnóstico, encontrándose los siguientes hallazgos microscópicos:
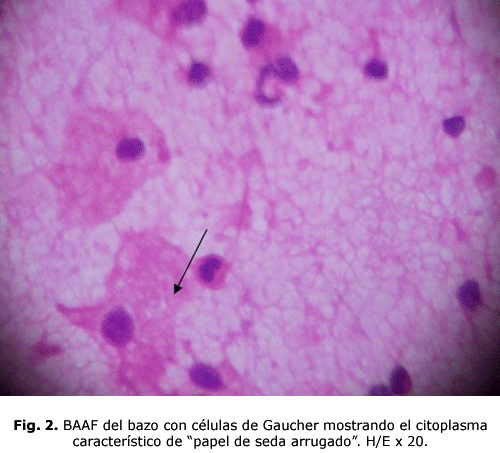
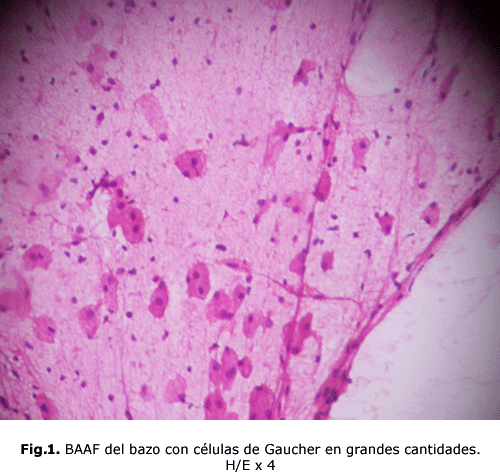
bazo: grandes células con citoplasma de tipo fibrilar que se ha comparado al papel de seda arrugado, conocidas como células de Gaucher. (fig. 1) y (fig. 2)
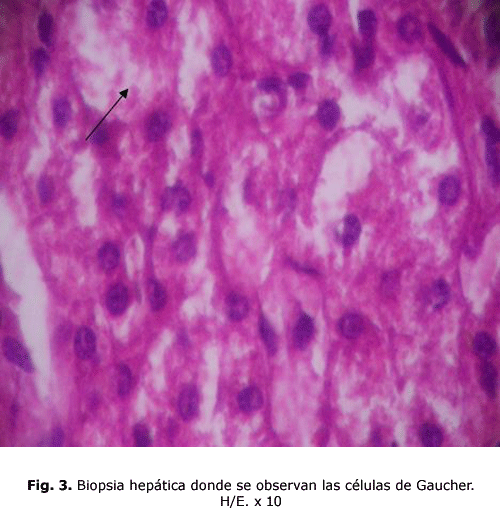
hígado: células de Gaucher entre los cordones de hepatocitos. (fig.3)
]]>
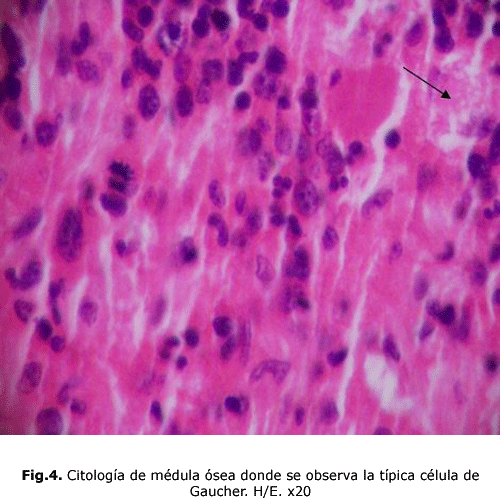
smear de médula ósea: la típica célula de Gaucher. (fig.4)
DISCUSIÓN
Las células de Gaucher por lo general son grandes y pueden medir hasta 100 micras de diámetro, pueden tener uno o más núcleos oscuros excéntricos, en raras ocasiones tienen vacuolas en su citoplasma y sí hay imagen de fibrillas. Con el microscopio electrónico se comprende que el aspecto fibrilar del citoplasma se debe a unos lisosomas alargados, estriados, cargados de lípidos que se depositan en doble capa formando pilas 5. Estas células son Ácido Periódico de Schiff (PAS), Sudán Black B con luz polarizada, Azul de Toluidina y Cresyl Violeta positivas. Se tiñen tenuemente con Oil Red o Red Sudán1.
Desde el punto de vista anatomopatológico al diferenciar esta lipidosis del resto de las enfermedades por depósito lisosomal, habría que descartar en primer lugar la Enfermedad de Wolman caracterizada también al igual que el caso que presentamos por la presencia de histiocitos con citoplasma espumoso cargado de lípidos situados en el intersticio, ya que el resto de este grupo de enfermedades como el Niemann Pick se caracterizan por presentar vacuolas lisosomales intracitoplasmáticas.
El diagnóstico se realizó basado en la clínica, fundamentalmente en la presencia de manifestaciones óseas y hepatoesplenomegalia. Se basó además en la observación histológica de las células de Gaucher y en la determinación de los niveles bajos de actividad de la enzima glucocerebrosidasa en los leucocitos de la sangre y fibroblastos cutáneos.
Células morfológicamente similares a las de Gaucher son encontradas en un pequeño número de enfermedades como son las leucemias mielógenas crónicas, leucemias agudas y anemias diseritropoyéticas.
REFERENCIAS BIBLIOGRÁFICAS
]]>1. Cotran RS, Kumar V, Collins T.Trastornos genéticos Robbins. En: Patología Estructural y Funcional. 6ta ed. Madrid: Mcgraw- Hill-Interamericana de España; 2000.p. 150-199.
2. Lee RE, Peters SP, Glew RH. Gaucher´s disease: clinical, morphologic, and pathogenic considerations. Pathol Annu. 1977; 12:309-339.
3. Fernandez J, Saudubray JM, Van den Berghe G, Walter JH. Inborn Metabolic Diseases. Diagnosis and Treatmet.4ta ed. Germany: Springer Medizin. 2006.
4. Pastores GM, Meer PA. Musculoskeletal complications associated with lysosomal storage disorders: Gaucher disease and Hurler-Scheie syndrome (mucopolysaccharidosis type I). Curr Opin Rheumtol. 2005[citado 23 mar 2016]; 17(1):70-78. Disponible en: http://ovidsp.tx.ovid.com/sp-3.25.0a/ovidweb.cgi?QS2
5. Weizman Z, Tennenbaum A, Yatziv S. Interphalangeal joint involvement in Gaucher’s disease, type I, resembling juvenile rheumatoid arthritis. Arthritis Rheum. 1982[citado 23 mar 2016]; 25(6):706-707. Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/art.1780250616/pdf
6. Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, et al. Enzyme replacement therapy and monitoring for children with type l Gaucher disease: consensus recommendations. J Pediatr. 2004[citado 23 mar 2016]; 144(1):112-120. Disponible en: http://www.sciencedirect.com/science/article/pii/S002234760300814X?via%3Dihub
7. Brady BO. The sphingolipidoses. N Engl J Med. 1966[citado 23 mar 2016]; 275(6):312-318. Disponible en: http://www.nejm.org/doi/full/10.1056/NEJM196608112750606
8. Adams WM. Neurohistochemistry. Amsterdan: Elsevier Publicating Company; 1965.
9. Amstuz H, Carey EJ. Skeletal manifestations and treatment of Gaucher's disease. Review of twenty cases. J Bone Joint Surg Am.1966; 48(4):670-701.
10. Anderson W, Kissane JM. Volume Two Pathology. Lincoln, United Kingdom: Published by The C. V. Mosby Company; 1977.
Recibido: 3 de febrero de 2017
Aprobado: 1 de marzo de 2017
]]>
Dra. Jenny María Patiño Pérez. Hospital General Vladimir Ilich Lenin. Holguín. Cuba.
Correo electrónico: jennym@infomed.sld.cu