REVISIÓN BIBLIOGRÁFICA
Mecanismos de inducción de la matriz extracelular en la nefropatía diabética
Mechanisms of induction of the extracellular matrix in diabetic nephropathy
Dra.C. Elizabeth Alejandrina Guzmán-Hernández, MSc. David Segura-Cobos
Facultad de Estudios Superiores Iztacala. Universidad Nacional Autónoma de México. México DF, México.
]]>
RESUMEN
La nefropatía diabética es una complicación grave en la diabetes mellitus. Sus principales cambios morfológicos típicos se deben al aumento de la cantidad de proteínas de la matriz extracelular. Los productos finales de glicación avanzada, resultado de la hiperglucemia, estimulan la producción de proteínas de la matriz extracelular en las células mesangiales, lo que resulta en la glomeruloesclerosis. Se revisan las alteraciones de las vías metabólicas que inducen la producción de factores que aumentan la síntesis de proteínas de la matriz extracelular y su acumulación durante el desarrollo de la nefropatía diabética. La glucosa intracelular elevada induce un aumento de angiotensina II y activación de proteína cinasa C, que a su vez, aumentan varios factores de crecimiento, como el transformante 1, el endotelial vascular, el de tejido conectivo, el epidérmico y el derivado de plaquetas, que llevan al incremento en la síntesis de proteínas de la matriz extracelular renal, como el colágeno, la fibronectina, la entactina y la laminina, lo que engrosará las membranas basales y expandirá progresivamente la matriz mesangial glomerular. Las metaloproteinasas de la matriz, que modulan la cantidad de proteínas de la matriz extracelular, son, a su vez, reguladas por los inhibidores tisulares de las metaloproteinasas.
Palabras clave: diabetes mellitus, nefropatía diabética, matriz extracelular, metaloproteinasas.
ABSTRACT
Diabetic nephropathy is a serious complication in diabetes mellitus. Its main and typical morphological changes are caused by the rise of the amount of the extracellulary matrix proteins. The final products of advanced glycation resulting from hyperglycemia stimulate the production of the extracellulary matrix proteins in the mesangial cells which leads to glomerulosclerosis. This article reviewed the alterations in the metabolic pathways that induce the production of factors capable of increasing the extracellulary matrix protein synthesis and their accumulation during the development of diabetic nephropathy. Elevated intracellular glucose leads to increased angiotensin II and C-kinase protein activation which, in turn, increase the number of several growth factors such as B1 transforming, vascular endothelial, connective tissue, epidermal and platelet-derived factors. All the above-mentioned causes more synthesis of renal extracellulary matrix proteins such as collagen, fibronectin, entactin and laminin which will thicken the basal membranes and will progresively extend the glomerular mesangial matrix. The matrix metalloproteins in charge of modulating the amount of proteins in the said matrix are then regulated by the tisular inhibitors of the metalloproteinases.
Keywords: diabetes mellitus, diabetic nephropathy, extracellulary matrix, metalloproteinases.
]]> INTRODUCCIÓN
Una de las complicaciones más relevantes de la diabetes mellitus (DM) es la nefropatía diabética (ND), que es un trastorno metabólico en el que la hiperglucemia y la consecuente glucosa intracelular elevada inducen disfunción en diversos tipos de células del riñón, que deriva en insuficiencia renal progresiva.1 En la actualidad, la ND representa casi el 50 % de los casos de enfermedad renal en etapa terminal.
La patogénesis de la DM 1 y 2 son distintas, pero los cambios que estos trastornos provocan en los glomérulos renales, el intersticio tubular renal y la vasculatura, son prácticamente indistinguibles.2 Por otra parte, parece que todos los tipos de células del riñón, incluyendo los podocitos, las células mesangiales, las células endoteliales y el epitelio tubular, se ven afectados por la elevada glucosa intracelular. Las etapas iniciales de la ND se pueden atribuir a la disfunción de los capilares glomerulares, y se manifiestan clínicamente por la presencia de hiperfiltración y microalbuminuria,3 debido a la disminución de la resistencia de las arteriolas aferente y eferente. Diversos factores bioquímicos, como las prostaglandinas, la angiotensina II (Ang II), el óxido nítrico (NO), el péptido natriurético atrial, el glucagón y la insulina, median estas alteraciones.3 En este trabajo se revisan las alteraciones de las vías metabólicas que inducen la producción de factores que aumentan la síntesis de proteínas de la matriz extracelular (MEC) y su acumulación durante el desarrollo de la ND.
Dentro de las alteraciones glomerulares características en la ND se encuentran la proliferación de las células mesangiales e hipertrofia, la acumulación excesiva de proteínas de la MEC en el mesangio y en la membrana basal glomerular, que finalmente conduce a la glomeruloesclerosis nodular, también conocida como lesión de Kimmelstiel-Wilson.4 Las alteraciones que se producen en el compartimiento tubulointersticial son similares, e incluyen la hipertrofia tubular, seguida del engrosamiento de la membrana basal tubular y fibrosis intersticial. Esto último puede deberse a un proceso único conocido como transformación mesenquimal epitelial, que transforma las células tubulares en células intersticiales; este proceso provoca una excesiva acumulación de proteínas de la MEC en el intersticio renal.4 Los alteraciones vasculares normalmente incluyen el engrosamiento y hialinización de las arteriolas aferentes y arterias interlobulares, por lo que se reduce la superficie disponible para la filtración y el estrechamiento u oclusión del lumen.4
DESARROLLO
Cambios en la matriz extracelular renal durante la nefropatía diabética
La MEC es una intrincada red macromolecular que se encuentra en el espacio extracelular, formado por proteínas (5 %) y proteoglicanos (95 %). La MEC se encuentra altamente hidratada, y en la interfase entre el epitelio y el tejido conjuntivo forma una lámina basal que participa en el control del comportamiento celular. Los principales componentes de la membrana basal del glomérulo son el colágeno tipo IV, la laminina y la entactina, mientras que los colágenos de los tipos I, V, VI y la fibronectina, se consideran generalmente como componentes del intersticio renal.5
En la ND las proteínas que se encuentran presentes en el mesangio son: los colágenos I, III y V, la fibronectina y la laminina; mientras que, en la lámina basal glomerular, se encuentran el colágeno IV y la entactina. Es claro que algunas proteínas como los colágenos I y III en las células mesangiales solo se presentan en las últimas etapas de la glomeruloesclerosis, asociadas con el desarrollo de nódulos Kimmelstiel-Wilson, en lugar de la expansión difusa de la MEC, que ocurre en las etapas tempranas, y moderadamente avanzadas de la enfermedad. La fibronectina se encuentra presente en las células mesangiales normales; sin embargo, en la ND aumenta su expresión y su síntesis.5
]]> Las proteínas de la MEC se someten normalmente a recambio metabólico, y son degradadas principalmente por las metaloproteinasas (MMPs),6 que son una familia de endopeptidasas dependientes de zinc, que desempeñan una gran variedad de procesos biológicos. Inicialmente se pensó que solo eran capaces de degradar proteínas de la MEC, pero hoy en día se conoce que pueden escindir una variedad de sustratos, que van desde los receptores de la superficie celular y moléculas de adhesión, a factores de crecimiento y citocinas. Todas las MMPs, con algunas modificaciones, consisten en un prodominio, un dominio catalítico, una región unión y un dominio de hemopexina.6Las MMPs se clasifican de acuerdo con su estructura y/o especificidad del sustrato en la MEC. Las colagenasas (MMP-1, MMP-8 y MMP-13) pueden degradar el colágeno y son antifibróticas. Las estromelisinas (MMP-3, MMP-10, y MMP-11) comparten una similitud estructural con las colagenasas, pero son incapaces de degradar el colágeno. Como su nombre indica, las gelatinasas (MMP-2 y MMP-9) escinden colágeno desnaturalizado (gelatina), así como el colágeno tipo IV en las membranas basales. Las matrilisinas (MMP-7 y MMP-26) degradan componentes de la MEC, como la laminina y la entactina.6
Alteraciones metabólicas en la diabetes mellitus
La exposición crónica a la glucosa sanguínea elevada tiene efectos perjudiciales sobre la secreción y síntesis de insulina, la supervivencia celular y la sensibilidad a la insulina a través de múltiples mecanismos de glucotoxicidad, lo que conduce a la hiperglucemia, y finalmente, al círculo vicioso de deterioro continuo de la función de las células beta.7 En oposición a la desensibilización de las células beta (un estado temporal de refractariedad celular a la estimulación de glucosa), o agotamiento de las células beta (agotamiento de las reservas de insulina reversibles con el reposo de las células), la glucotoxicidad implica cambios irreversibles de los componentes celulares de la secreción y producción de insulina.7
Existen múltiples vías y mecanismos mediante los cuales la hiperglucemia crónica puede alterar la función y provocar la apoptosis de las células beta, dentro de las cuales se encuentran:
]]>- La exposición prolongada al aumento en la concentración de glucosa causa pérdida gradual de la expresión de gen de la insulina, secundaria a la disminución de la actividad de factores de transcripción y otros genes específicos de las células beta.8
- Las células beta, cuando son expuestas a un aumento de la secreción de insulina, inducen gran demanda en el retículo endoplásmico (RE) para la síntesis de la proinsulina, que deriva en estrés celular. El estrés en el RE consiste en la acumulación de proteínas desplegadas; cuando se genera un estrés crónico, se induce a la muerte celular programada de las células beta mediada por cinasas de estrés y factores de transcripción.8
- La hiperglucemia sostenida por un largo plazo aumenta el flujo metabólico en las mitocondrias, e induce la generación excesiva de especies reactivas de oxígeno (EROs), que dan lugar a estrés oxidativo crónico.9 Estudios clínicos indican que los pacientes diabéticos están expuestos a estrés oxidativo crónico y presentan un incremento de pro-oxidantes y marcadores de daño tisular oxidativo. Por otra parte, estudios realizados en islotes de páncreas aislados de pacientes con DM 2 mostraron un aumento en los marcadores de estrés oxidativo, y estos se correlacionaron con el deterioro de la secreción de insulina estimulada por la glucosa. Durante la hiperglucemia, la producción excesiva de las EROs se genera por la fosforilación oxidativa mitocondrial durante la glicólisis anaeróbica y de vías alternas, en las que la glucosa se desvía cuando se excede la capacidad glucolítica hacia la autooxidación de la glucosa, la glicación no enzimática, la activación de la proteína cinasa C, la hexosamina y el sorbitol.9
Durante la hiperglucemia crónica la glucosa puede interaccionar con otros carbohidratos, como la fructosa y la glucosa-6-fosfato, o sus derivados con las proteínas, ácidos nucleicos y lípidos, para formar productos finales de glicación avanzada, conocidos como AGEs (por sus siglas en inglés, advanced glycation end products).10 Este proceso, es conocido como glicación, está asociado con el envejecimiento que se acelera con la diabetes, y se inicia con la reacción de los grupos carbonilos de los carbohidratos con los grupos amino de las proteínas, en especial, con el amino terminal y el ε-amino de residuos de lisina, dando origen a los productos tempranos de glicación, también llamados de Amadori o fructosamina. A partir de ellos, y por transposiciones moleculares y oxidaciones, se forman compuestos α-dicarbonilos (α-oxoaldehídos) como la 3-desoxiglucosona, el metilglioxal y el glioxal, que son conocidos como precursores de los productos finales de glicación avanzada (AGEs); al combinarse simultáneamente con las proteínas, forman puentes cruzados muy estables entre ellas, que producen su agregación y pérdida de sus funciones biológicas. Las proteínas ricas en aminoácidos básicos (L-lisina y L-arginina) son especialmente susceptibles a la glicación.10
La 3-desoxiglucosona también se puede formar directamente por la autooxidación de la glucosa catalizada por metales, proceso en el cual se produce el radical superóxido, precursor de otras especies reactivas de oxígeno.
El metilglioxal y el glioxal derivan principalmente de intermediarios de la glucólisis como el gliceraldehído-3-fosfato y la dihidroxiacetona-fosfato, que se acumulan a consecuencia de la hiperglucemia. Las concentraciones de metilglioxal y 3-desoxiglucosona se elevan en el plasma del modelo animal con hiperglucemia y en el paciente diabético.11
La formación intracelular de AGEs es más rápida que la extracelular, por lo que se ha propuesto que la alta concentración intracelular de glucosa desencadena la producción de los AGEs intra y extracelularmente.11 Diversos estudios han mostrado que la formación de los AGEs aumenta en proporción a la glucemia, hecho que sugiere que elevaciones moderadas resultan en la acumulación sustancial de estos AGEs en los pacientes diabéticos. Períodos cortos de hiperglucemia, como ocurre con las alteraciones de la tolerancia a la glucosa, pueden ser suficientes para fomentar su formación.11
Las proteínas modificadas por los AGEs pueden encontrarse en el plasma, en el compartimiento intracelular y en la MEC; especialmente, en la pared arterial, en el mesangio glomerular, en la membrana basal del glomérulo, en los vasos capilares sanguíneos, en la vasculatura de la retina, el cristalino y las fibras nerviosas mielínicas y amielínicas.12 Además, se ha encontrado que los AGEs inhiben la actividad de las MMP, lo que permite que se acumulen en el espacio extracelular. Por otra parte, la glicación de proteoglicanos sulfatados reduce su electronegatividad, y por lo tanto, modifica la selectividad de la membrana basal glomerular, lo que resulta en la microalbuminuria.12
Los AGEs, a través de su unión a su receptor (RAGE), participan en los cambios de la adhesividad de la célula, en la hiperpermeabilidad de los capilares y en las interacciones célula-matriz.13 Las proteínas glicadas que se unen a estos receptores inducen diversos eventos, como la producción de EROs, y la activación de factores de transcripción como el NF-kB, así como la expresión de diversos péptidos y proteínas como factores de crecimiento y citocinas, como el factor de crecimiento transformante β1 (TGF-β1) y el factor de necrosis tumoral alfa (TNF-α) que estimulan la síntesis de proteínas de la MEC.13
Incremento en la actividad de la vía del sorbitol
Efectos de la acumulación de la fructosa
La transformación bioquímica de la fructosa que afecta al organismo se inicia en la mayoría de los tejidos, excepto en el hígado, con su fosforilación por una hexocinasa específica, y da lugar a la fructosa-6-fosfato, que se metaboliza en la ruta glucolítica. De esta manera, la vía de los polioles y la glucólisis se relacionan y conducen a diversas alteraciones metabólicas, entre las que se encuentran: la acumulación de intermediarios de la glucólisis, principalmente de gliceraldehido-3-fosfato (G3P) y la dihidroxiacetona-fosfato (DHAP), ambos altamente reactivos con la capacidad de glicar proteínas y generar estrés oxidativo.16
La enzima gliceraldehído-3-fosfato-deshidrogenasa (G3PDH) desempeña un papel fundamental en la génesis de las complicaciones diabéticas. Cuando la concentración intracelular de la glucosa es alta, su actividad es inhibida, razón por la cual se acumulan las triosas fosfato.
Otro compuesto derivado de la fructosa es la fructosa-3-fosfato, la cual se acumula en los cristalinos y eritrocitos de ratas diabéticas. Cuando se hidroliza este producto, produce 3-desoxiglucosona, y ambos tienen la propiedad de glicar proteínas. Es evidente que el aumento de la fructosa-3-fosfato y su hidrólisis contribuyen a la glicación de proteínas, efecto que depende de la concentración de la 3-desoxiglucosona, de la velocidad de hidrólisis de la fructosa-3-fosfato y de la eficiencia de los mecanismos de desintoxicación para la 3-desoxiglucosona.16
Activación de la vía de las hexosaminas
La fructosa vía sorbitol contribuye en la activación de la vía de las hexosaminas, debido a que la formación de la glucosamina-6-fosfato proviene, exclusivamente, de la fructosa-6-fosfato y la glutamina, mediante una reacción irreversible catalizada por la glutamina: fructosa-6-fosfato amido transferasa (GFA), enzima que regula la vía. La glucosamina-6-fosfato a través de reacciones subsecuentes, origina la UDP-N-acetilglucosamina y la UDP-N-acetilgalactosamina, que se utilizan en la formación de las glicoproteínas y proteoglicanos.16
El aumento del flujo a través de esta vía está relacionado con la estimulación de la expresión de genes como los del TGF-α, TGF-β y del inhibidor del activador del plasminógeno-1 (PAI-1), que deriva en la acumulación de proteínas de la MEC.16
]]>La acumulación de dihidroxiacetona-fosfato y gliceraldehido-3-fosfato promueve la producción de diacilglicerol (DAG), y la subsecuente activación de la proteína cinasa C (PCC), los mismos que también afectan la homeostasis vascular.17 La PCC juega un papel central en la ND, existen varias isoformas dentro de las cuales se encuentran la α, β, δ, ε y ξ, que se expresan en el riñón, y se activan en ratas diabéticas y en cultivos celulares de glomérulos.17 La PCC es activada por múltiples rutas, principalmente DAG, que se forma a partir de la dihidroxiacetona-fosfato, o bien la PCC se puede activar a partir del aumento de la actividad de la vía del poliol, la glucosamina y las EROs.17
Las alteraciones celulares y funcionales atribuidas a la activación de la PCC son muy variadas, y dependen de la función de esta enzima en los mecanismos de transducción de señales. Por ejemplo, la PCC participa en la inducción de la expresión de genes de proteínas de la MEC como la fibronectina y colágeno tipo IV.17
Factores de crecimiento y hormonas que afectan la expresión de proteínas de la matriz extracelular en la nefropatía diabética
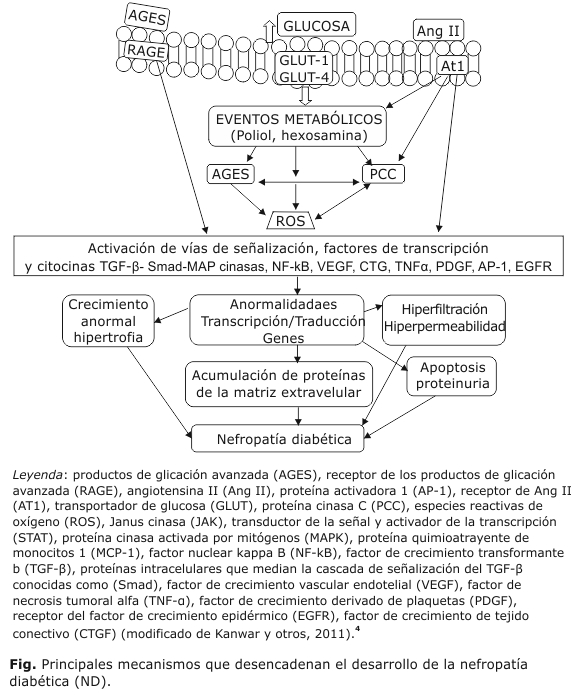
El balance entre la formación y la degradación de la MEC se encuentra regulada por factores de crecimiento como el TGF-β, el factor de crecimiento de tejido conectivo (CTGF), el factor de crecimiento semejante a la insulina (IGF-1), el factor de crecimiento fibroblástico, el factor de crecimiento epidérmico (EGFR), y el factor de crecimiento derivado de plaquetas (PDGF) (Fig.).
Angiotensina II
La Ang II es un octapéptido vasoactivo, que al unirse con su receptor AT1, induce a que la proteína Gq/11 active a la fosfolipasa C, que hidroliza fosfatidilinositol bifosfato y produce inositol trifosfato y DAG. DAG estimula a la PCC, que induce un aumento en la síntesis y actividad del transportador de glucosa GLUT1, lo que resulta en aumento del flujo y utilización de glucosa, que desencadena la producción de más DAG y activación adicional de la PCC,18 por lo que se induce la expresión de factores de transcripción como AP-1, el cual tiene un sitio de reconocimiento en el promotor del gen de la fibronectina, e induce, de esta manera, mayor síntesis de esta proteína de la MEC.18
]]> La Ang II también a través de su unión al receptor AT1 induce la fosforilación de otras cinasas de tirosinas, como non receptor protein-tyrosine kinase (Src) y la proteína (Smad1), que inducen el aumento de la síntesis de colágeno IV en las células mesangiales,19 o también puede inducir la fosforilación de otros factores de transcripción como el ETS-1 a través de la activación y fosforilación de la vía de señalización ERK 1/2 o Akt/PKB, que estimulan mayor producción de fibronectina en las células mesangiales.20
Hormona de crecimiento
La hormona de crecimiento (GH), es un polipéptido de 191 aminoácidos secretada por la glándula pituitaria anterior, que está bajo el control de factores hipotalámicos de GHRH y la somatostatina. GH estimula la síntesis del factor de crecimiento semejante a la insulina (IGF-1) en varios órganos a través de la unión con su receptor (GHR). GH afecta el metabolismo de grasas, proteínas y carbohidratos, acciones que son posibles debido a su receptor GHR. La porción extracelular de GHR se escinde, y se une a una proteína de unión llamada GHBP. La unión de la hormona de crecimiento a su receptor GHR estimula la activación de varias vías de señalización, como la cinasas de tirosina Janus cinasa 2 (JAK2), proteínas transductoras de señales y activadoras de la transcripción (STAT), proteínas cinasas activadas por mitógeno (MAPK) y la fosfoinosítido 3 cinasa (PI3-K).21
Los podocitos son altamente ramificados y diferenciados en las células epiteliales terminales, son críticos para la formación de la barrera de filtración glomerular que evita la pérdida de macromoléculas, como las proteínas en el filtrado glomerular. El daño y la pérdida de los podocitos es una alteración temprana y fundamental de la ND en los seres humanos.21 Existe una relación directa entre la actividad de GH/IGF-1 y la hipertrofia renal.
La DM se asocia con una disminución de podocitos. En ratones transgénicos de GH se observaron alteraciones significativas de la barrera de filtración, que se caracterizan por la inducción de la cinasa vinculada a integrinas (ILK), que altera la función de los podocitos a través de la inhibición de la expresión de E-cadherina y aumento del complejo de transcripción b-catenina/LEF, DNA-bound/lymphoid enhancer factor con aumento consecutivo de la metaloproteinasa-9, que es una gelatinasa que degrada colágeno IV, crucial para el mantenimiento de la membrana basal glomerular. Niveles incrementados de GH en los podocitos activa a otros factores de transcripción, como el Sip1, que inhibe la expresión de E-cadherina, que es una proteína podocitaria que forma parte del diafragma de filtración. Esto lleva a que aumente la permeabilidad y el paso de proteínas a través de la monocapa de podocitos durante la ND.21
Factor de crecimiento transformante β
El factor de crecimiento transformante β (TGF-β), es una citocina pro-esclerótica directamente involucrada en la patogénesis de la glomeruloesclerosis y la fibrosis intersticial. Es un prototipo de la superfamilia de TGF, que ejerce efectos pleiotrópicos, es decir, inhibe la proliferación y la apoptosis, pero induce hiperplasia e hipertrofia de las células mesangiales.20 En la MEC, existe TGF-β1 como una forma latente, formando un complejo con las proteínas plasmina o trombospondina 1 (TSP-1). La hiperglucemia aumenta la expresión de TSP-1, la cual interactúa específicamente con el TGF-β latente para activarlo.21
El TGF-β1 activo inicia sus efectos celulares mediante la unión a su receptor tipo III, posteriormente forma un ligando con receptor tipo II, tras la formación del complejo, el receptor tipo II fosforila residuos de serina y treonina en el receptor de tipo I, activando de esta manera su actividad cinasa, lo que conduce a la activación de factores de transcripción como Smad 2 o Smad 3, los cuales se translocan al núcleo para regular la transcripción de genes como el del colágeno I, PAI-1, la c-Jun y la fibronectina.21
]]> Las cinasas activadas por señales extracelulares 1 y 2 (ERK1/2), la cinasa p44/MAPK p42, la cinasa c-Jun N-terminal/activada por estrés de la proteína cinasa (JNK/SAPK), y MAP cinasa p38 junto con las Smads, regulan la actividad del TGF-β1 en las células mesangiales. Estas cinasas regulan la transcripción del procolágeno y de la fibronectina a través de AP-1.22La acumulación de la MEC también puede estar relacionada con la inhibición de MMPs y la activación de inhibidores tisulares de las MMP (TIMP). La expresión de genes de proteínas de la MEC también es modulada por otra citocina, el factor CTGF, que es inducido por el TGF-β1 a través de la inhibición del factor de transcripción TIEG-1, y perpetúa la activación de la señalización del TGF-β.23
La administración de anticuerpos neutralizantes anti-TGF-β1 evita la hipertrofia renal, la expansión de la matriz mesangial, el aumento de colágeno IV y de fibronectina, en ratones con diabetes inducida por estreptozotocina, lo que sugiere un papel potencial para el TGF-β1 en la patogénesis de la ND.24
En ratones, la coinyección subcutánea de TGF-β y CTGF produce fibrosis sostenida y persistente. En varios modelos experimentales, como la obstrucción unilateral del uréter, la nefritis por anticuerpos anti-Thy1 y la infusión de Ang II, TGF-β y CTGF se encuentran aumentados en etapas avanzadas de fibrosis, lo cual indica que estos factores contribuyen a la progresión del daño renal.25
La expresión de algunas proteínas de la MEC como la fibronectina, que es dependiente de CTGF, en las células mesangiales, está mediada por la activación de la vía de señalización MAPK y PKB, mientras que el colágeno I, PAI-1 y TIMP-1 son dependientes de TGF-β.25
Factor de crecimiento endotelial vascular
El factor de crecimiento endotelial vascular (VEGF-A) mantiene la integridad y viabilidad del endotelio, su diferenciación y supervivencia, a través de la proliferación de las células endoteliales; es altamente expresado por los podocitos, y es importante en el desarrollo glomerular.26
El VEGF-A puede promover el desarrollo de la ND a través de un desacoplamiento con el NO. El VEGF provoca la liberación del NO, y actúa como factores tróficos en el endotelio. El NO derivado de las células endoteliales evita la proliferación de células endoteliales y del músculo liso, y la infiltración de macrófagos. En los ratones con deleción génica (knockout) para sintasa de óxido nítrico (NOS), el aumento en la expresión de VEGF se asoció con el desarrollo de glomeruloesclerosis y sobreexpresión del transportador de glucosa (GLUT1) en las células mesangiales.27
El VEGF-A induce la proliferación mesangial, estimula la activación del TGF-β y la síntesis de colágeno a3 IV por los podocitos y las células mesangiales. La glucosa alta y el VEGF-A inducen la activación rápida del TGF-β latente en TGF-β activo, mediada por las metaloproteinasas (MMP-2 y MMP-9), estimulando el crecimiento celular y la proliferación celular. La señalización celular estimulada por el TGF-β1 se considera responsable para la proliferación de las células mesangiales y la expansión de la MEC observada en la ND en humanos y roedores.27
]]> El VEGF-A y el TGF-β inducen la expresión del CTGF, que es una proteína secretada matricelular modular, rica en cisteína, de la familia CCN y mediadores del TGF-β, que es crítico para el desarrollo de la fibrosis. El CTGF es mitogénico para las células endoteliales y mesangiales, e induce la síntesis de colágeno IV y fibronectina proteínas de la MEC.27
Factor de crecimiento epidérmico
El factor de crecimiento epidérmico (EGF) y su receptor EGFR es una glicoproteína transmembranal con actividad de tirosina cinasa que influye en la proliferación, la diferenciación y la tumorigénesis. En la DM, el EGFR puede modular el engrosamiento de la pared vascular y las complicaciones microvasculares de la enfermedad.28 En este sentido, el remodelado de la pared vascular en ratones diabéticos se mejora tras la inhibición de la tirosina cinasa. El EGFR puede ser activado por varios ligandos, incluyendo el EGF, y EGF con heparina unida (HB-EGF). HB-EGF se libera de la superficie de la célula por escisión enzimática.28
Recientemente, se ha hecho evidente que el EGFR no solo media la proliferación celular a través de unión a ligando, sino que también la transactivación de EGFR juega un papel fundamental en la facilitación de la señalización intracelular inducida por la interacción con la Ang II y su unión al receptor AT1. La transactivación se ha descrito en las células epiteliales tubulares, en células mesangiales, e incluso, en los podocitos, por lo que puede representar un mecanismo común que une la hipertrofia renal asociada a la disminución progresiva del funcionamiento renal.28
La glucosa intracelular elevada en las células mesangiales activa a la cinasa de proteínas en tirosina Src (oncoproteína viral activada durante el sarcoma de Rous), lo cual conduce a la transactivación del EGFR, seguido de la fosforilación de las MAP cinasas y síntesis de colágeno IV.29
Otra vía de activación es la PI3/Akt, que a través del EGFR, induce la síntesis de colágeno I en las células mesangiales.30
Factor de crecimiento derivado de plaquetas
El factor de crecimiento derivado de plaquetas (PDGF) se expresa en las células mesangiales. Cuando el PDGF se une a su receptor PDGFR induce la autofosforilación del dominio citoplasmático de tirosina cinasa PDGFR y posterior reclutamiento de proteínas adaptadoras de entre 55 y 70 aminoácidos, identificado por primera vez como una región conservada en la proteína viral v-Crk llamadas SH2 y SH3. Estas proteínas inducen vías de señalización, como la activación de la vía de la fosfolipasa C, que resulta en la expresión de genes implicados en la proliferación celular y deposición de proteínas de la MEC,31 por lo que su expresión se incrementa durante la ND.
]]> En estudios realizados en pacientes y en animales diabéticos se ha observado un aumento en la expresión del PDGF en el glomérulo, seguido de la acumulación de proteínas de la matriz tubulointersticial, cambios que son atenuados tras la administración de un inhibidor de la tirosina cinasa.31
Factor de necrosis tumoral alfa
El factor de necrosis tumoral alfa es una citocina con efectos proinflamatorios, que se produce principalmente por los monocitos, macrófagos y células T, pero también en células renales intrínsecas. TNF-α puede causar citotoxicidad directa en las células renales, inducir apoptosis y muerte celular necrótica; como también puede producir alteraciones del flujo sanguíneo intraglomerular, reducción de la tasa de filtración glomerular y cambios en la permeabilidad de las células endoteliales.32
El TNF-α es capaz de inducir directamente la formación de EROs por las células renales. Investigaciones recientes han demostrado en glomérulos aislados de ratas que el TNF-α induce la activación de la NADPH oxidasa a través de la activación de la PCC y las MAP cinasas, resultando en alteraciones de la pared capilar glomerular.32 Estudios realizados in vitro muestran que el TNF-α estimula la absorción de solutos en las células tubulares proximales secundaria a la activación de cotransportadores dependientes de sodio, mientras que estudios in vivo en ratas diabéticas mostraron aumento de la excreción urinaria de TNF-α, que fue asociada a la retención de sodio y a la hipertrofia renal. Otros estudios han mostrado que la expresión de TNF-a en la corteza renal y su excreción urinaria se correlaciona con la presencia dealbuminuria.33 TNF-α estimula la secreción de la metaloproteinasa de matriz 9 (MMP-9), la cual interrumpe la integridad celular del podocito y promueve la permeabilidad a la albúmina de la monocapa de podocitos y la síntesis de proteínas de la MEC.33
CONCLUSIONES
Los factores de crecimiento transformante β1 y de necrosis tumoral α y los AGEs, son los principales inductores de la síntesis y acumulación de las proteínas de la MEC que conducen a la progresiva expansión de la matriz mesangial y el engrosamiento de la membrana basal glomerular durante la fisiopatología de la ND. La comprensión de las vías bioquímicas que estimulan la expresión de proteínas de la matriz podría conducir al desarrollo de nuevas estrategias de intervención en la ND.
REFERENCIAS BIBLIOGRÁFICAS
]]>1. Kolset SO, Reinholt FP, Jenssen T. Diabetic Nephropathy and Extracellular Matrix. J Histochem Cytochem. 2012;60(12):976-86.
2. Fioretto P, Mauer M. Histopathology of diabetic nephropathy. Semin Nephrol. 2007;27:195-207.
3. Wolf G, Ziyadeh FN. Cellular and molecular mechanisms of proteinuria in diabetic nephropathy. Nephron Physiol. 2007;106(2):26-31.
4. Kanwar YS, Sun L, Xie P, Liu FY, Chen S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol. 2011;6:395-423.
5. Mariappan MM. Signaling mechanisms in the regulation of renal matrix metabolism in diabetes. Exp Diabetes Res. 2012;2012:749812. Epub 2012 Feb 19.
]]>6. Morrison CJ, Butler GS, Rodriguez D, Overall CM. Matrix metalloproteinase proteomics: substrates, targets, and therapy. Curr Opin Cell Biol. 2009;21:645-53.
7. Maris M, Ferreira GB, D'Hertog W, Cnop M, Waelkens E, Overbergh L, Mathieu C. High glucose induces dysfunction in insulin secretory cells by different pathways: a proteomic approach. J Proteome Res. 2010;9(12):6274-87.
8. Flamment M, Foufelle F. Endoplasmic reticulum stress: from physiology to pathogenesis of type 2 diabetes. MedSci (Paris). 2013;29(8-9):756-64.
9. Wu Y, Tang L, Chen B. Oxidative Stress: Implications for the Development of Diabetic Retinopathy and Antioxidant Therapeutic Perspectives. Oxid Med Cell Longev. 2014;2014:1-12.
10. Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014;2:411-29.
]]>11. Singh VP, Bali A, Singh N, Jaggi AS. Advanced Glycation End Products and Diabetic Complications. Korean J Physiol Pharmacol. 2014;18(1):1-14.
12. Schalkwijk CG, Miyata T. Early-and advanced non-enzymatic glycation in diabetic vascular complications: the search for therapeutics. Amino Acids. 2012;42(4):1193-204.
13. Manigrasso MB, Juranek J, Ramasamy R, Schmidt AM. Unlocking the Biology of RAGE in Diabetic Microvascular Complications. Trends Endocrinol Metab. 2014;25(1):10.
14. Chiu CJ, Taylor A. Dietary hyperglycemia, glycemic index and metabolic retinal diseases. Prog Retin Eye Res. 2011;30(1):18-53.
15. Maccari R, Ottanà R. Targeting aldose reductase for the treatment of diabetes complications and inflammatory diseases: new insights and future directions. J Med Chem. 2015;58(5):2047-67.
]]>16. Kitada M, Zhang Z, Mima A, King GL. Molecular mechanisms of diabetic vascular complications. J Diabetes Investig. 2010;1(3):77-89.
17. Thallas-Bonke V, Cooper ME. Tandem Inhibition of PKC in Diabetic Nephropathy: It Takes Two to Tango? Diabetes. 2013;62(4):1010-11.
18. Heilig CW, Deb DK, Abdul A, Riaz H, James LR, Salameh J, Nahman NS Jr. GLUT1 regulation of the pro-sclerotic mediators of diabetic nephropathy. Am J Nephrol. 2013;38(1):39-49.
19. Mima A, Matsubara T, Arai H, Abe H, Nagai K, Kanamori H, et al. Angiotensin II-dependent Src and Smad1 signaling pathway is crucial for the development of diabetic nephropathy. Lab Invest. 2006;86:927-39.
20. Kumar PA, Brosius FC 3rd, Menon RK. The glomerular podocyte as a target of growth hormone action: implications for the pathogenesis of diabetic nephropathy. Curr Diabetes Rev. 2011;7(1):50-5.
]]>21. Lan HY, Chung AC. TGF-β/Smad signaling in kidney disease. Semin Nephrol. 2012;32(3):236-43.
22. Kanwar YS, Sun L, Xie P, Liu FY, Chen S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol. 2011;6:395-423.
23. McLennan SV, Abdollahi M, Twigg SM. Connective tissue growth factor, matrix regulation, and diabetic kidney disease. Curr Opin Nephrol Hypertens. 2013;22(1):85-92.
24. Garud MS, Kulkarni YA. Hyperglycemia to nephropathy via transforming growth factor beta. Curr Diabetes Rev. 2014;10(3):182-9.
25. McLennan SV, Abdollahi M, Twigg SM. Connective tissue growth factor matrix regulation and diabetic kidney disease. Curr Opin Nephrol Hypertens. 2013;22:85-92.
]]>26. Advani A. Vascular endothelial growth factor and the kidney: something of the marvellous. Curr Opin Nephrol Hypertens. 2014;23(1):87-92.
27. Tufro A,Veron D. VEGF and podocytes in diabetic nephropathy. Semin Nephrol. 2012;32(4):385-93.
28. Advani A, Wiggins KJ, Cox AJ, Zhang Y, Gilbert RE, Kelly DJ. Inhibition of the epidermal growth factor receptor preserves podocytes and attenuates albuminuria in experimental diabetic nephropathy. Nephrology. 2011;16(6):573-81.
29. Taniguchi K, Xia L, Goldberg HJ, Lee KW, Shah A, Stavar L, et al. Inhibition of Src kinase blocks high glucose-induced EGFR transactivation and collagen synthesis in mesangial cells and prevents diabetic nephropathy in mice. Diabetes. 2013;62(11):3874-86.
30. Wu O, Peng F, Zhang B, Ingram AJ, Gao B, Krepinsky JC. Collagen I induction by high glucose levels is mediated by epidermal growth factor receptor and phosphoinositide 3-kinase/Akt signaling in mesangial cells. Diabetologia. 2007;50(9):2008-18.
]]>31. Boor P, Ostendorf T, Floege J. PDGF and the progression of renal disease. Nephrol Dial Transplant. 2014;29:i45–i54.
32. García PM, Getino MA, Domínguez V, Navarro JN. Inflammation in diabetic kidney disease. World J Diabetes. 2014;5(4):431-43.
33. Li SY, Huang PH, Yang AH, Tarng DC, Yang WC, Lin CC, et al. Matrix metalloproteinase-9 deficiency attenuates diabetic nephropathy by modulation of podocyte functions and dedifferentiation. Kidney Int. 2014;86(2):358-69.
Recibido: 12 de marzo de 2015.
Aprobado: 4 de mayo de 2015.
]]>
Elizabeth Alejandrina Guzmán Hernández. Facultad de Estudios Superiores Iztacala. Universidad Nacional Autónoma de México. Avenida de los Barrios 1, Colonia Reyes Iztacala Tlalnepantla. CP 54090. Estado de México, México. Correo electrónico: shponia2000@yahoo.com.mx
]]>