Hiperplasia suprarrenal congénita por déficit de 21-hidroxilasa. Presentación de dos casos
Congenital adrenal hyperplasia related to hydroxilase-21 deficiency. A two-case report
José Rafael Hernández Gómez1, Charlia Preciado Delgado2, Olga Lidia Pérez Álvarez3, Jesús Juan Rodríguez4.
]]>
1 Especialista de I Grado en Endocrinología. Asistente. Hospital Pediátrico "Pepe Portilla". Pinar del Río.
2 Especialista de I Grado en Pediatría. Instructora. Hospital Pediátrico "Pepe Portilla". Pinar del Río.
3 Especialista de I Grado en Pediatría. Instructora. Hospital Pediátrico "Pepe Portilla". Pinar del Río.
4 Especialista de II Grado en Pediatría y Genética Clínica. Asistente. Hospital Pediátrico "Pepe Portilla". Pinar del Río.
RESUMEN
Se presentan dos recién nacidos con Hiperplasia Suprarrenal Congénita por déficit de 21-hidroxilasa clásica, diagnosticadas en el Servicio de Endocrinología del Hospital Pediátrico Provincial Docente "Pepe Portilla" de Pinar del Río, Cuba. Se exponen las diferencias clínicas entre los niños afectados en relación con el sexo, evidenciando dificultades diagnósticas en el varón, que lo expone a complicaciones graves por los desequilibrios hidroelectrolíticos que pueden sufrir con elevado riesgo para su vida. Se presenta información sobre la entidad, revisando la genética de la misma y algunos adelantos en su diagnóstico precoz. ]]>
Palabras clave: Hiperplasia SUPRARRENAL congénita-ambigüedad genital-Pérdida salina - diagnóstico NEONATAL.
ABSTRACT
Two infants presenting congenital adrenal hyperplasia due to classical 21-hydroxylase deficiency were diagnosed in Endocrinology Service belonging to "Pepe Portilla" Pediatric Teaching Provincial Hospital, Pinar del Río, Cuba. Clinical differences among the infants affected in relation to sex were showed, making evident diagnostic difficulties in male infant, which increase the risk of severe electrolyte imbalance complications for life. Information about the entity was presented, revising its genetics and some advances for its early diagnosis.
Key words: CONGENITAL ADRENAL HYPERPLASIA, GENITAL AMBIGUITY, SALT DEPLETION, NEONATAL DIAGNOSIS.
INTRODUCCIÓN
La Hiperplasia Suprarrenal Congénita (HSC) comprende un grupo de trastornos autosómicos recesivos que comprometen la síntesis del cortisol y otros esteroides adrenales, provocando un aumento compensatorio de la hormona adrenocorticotropa (ACTH), hiperplasia adrenal y acumulación de metabolitos anteriores al bloqueo metabólico. La expresión fenotípica dependerá de la naturaleza y severidad del defecto enzimático para la síntesis del cortisol y/o aldosterona.1 ]]>
El déficit de 21-hidroxilasa es la causa más frecuente de Hiperplasia Suprarrenal Congénita, caracterizada clínicamente por signos de hiperandrogenismo y déficit mineralocorticoide.1,2 Se trata de una enfermedad hereditaria autosómica recesiva, determinante del déficit de la enzima 21-hidroxilasa del citocromo enzimático P450c21, codificada por el gen funcional CYP21 (o CYP21B) identificado en 1985, localizado en el Complejo Mayor de Histocompatibilidad HLA, mapeado su locus en la región 6p21, 3 del brazo corto del cromosoma 6, que se dispone en tándem con un gen inactivo llamado CYP21P (seudo gen CYP21A).2-4 Ambos genes se encuentran adyacentes a los genes C4A y C4B, que codifican para el cuarto componente del complemento; el orden de los genes es C4A-CYP21A-C4B-CYP21B.3,4 Por estudios de caracterización molecular en pacientes con diferentes tipos de deficiencia de 21-hidroxilasa se identificaron varias mutaciones: a) de lesiones en estado homocigoto del gen CYP21B en la forma clásica perdedora de sal, y b) pequeños intercambios de secuencia entre los genes CYP21A y CYP21B, fenómeno denominado "conversión génica", que conducen a la transferencia de una de las mutaciones del seudo gen al gen funcional; además se han identificado varios polimorfismos normales en el gen CYP21B, los cuales no afectan la actividad de 21- hidroxilasa.5,6 La observación de que el gen de la 21-hidroxilasa segrega junto con los determinantes antigénicos del complejo HLA-B sugirió el estudio de haplotipos de CMH en familias afectadas con la deficiencia de 21-hidroxilasa, lo que ha permitido establecer que cualquier miembro de una familia que comparta un haplotipo con el sujeto afectado, es portador del gen del defecto enzimático.2,3Existen dos formas de expresión de la enfermedad:
a) la forma clásica, que puede ser completa (virilizante y perdedora de sal) o incompleta (virilizante simple).
b) la forma no clásica, que puede ser sintomática o no, donde el déficit enzimático, por lo general moderado, cursa con ausencia de ambigüedad genital al nacimiento y signos de virilización posterior (adrenarquia o seudo pubertad precoz, hirsutismo, acné, irregularidades menstruales e infertilidad).1,7
La incidencia de las formas clásicas es de 1/15.000 nacimientos8 y en las formas no clásicas de 1/500-1/1.000 en población de raza blanca.9
Desde el punto de vista clínico humoral el diagnóstico del déficit de 21-hidroxilasa está determinado por el incremento del precursor del cortisol 17-OH-progesterona en presencia de virilización acompañada o no de pérdida de sal (deshidratación, hiponatremia e hiperpotasemia).1
Caso 1.- P.M.R.S.
Antecedentes prenatales: Malformación congénita de extremidades en abuelo materno. Cariotipo prenatal: 46, XY. Ultrasonido a las 29 semanas de gestación con aumento de tamaño de ambos riñones fetales y ecogenicidad aumentada. Senescencia precoz de la placenta. No consanguinidad parental.
Antecedentes perinatales: Parto eutócico e institucional a término, raza blanca y primogénito, con peso de 3940 gramos y talla de 51 cm.
Ingresa a los 13 días de edad en el Servicio de Neonatología con pérdida del 11.1% del peso corporal (peso: 3500 gramos) y poca avidez por la leche, asociándose además síntomas respiratorios altos y febrícula; se interpretó como un Catarro Común. El día 15 se encuentra que no progresa de peso, ha comenzado con regurgitaciones postprandiales y su succión es débil, el cuadro clínico se interpretó como una Sepsis del Recién Nacido, los complementarios exhiben leucocitosis a predominio linfocitario con eritrosedimentación discretamente acelerada (30 mm/h) y acidosis metabólica ligera (BE menor de 7 mEq/L). Se realizó una radiografía de tórax en la que se observaron lesiones parenquimatosas que se interpretan como una Bronconeumonía Bacteriana trasladándose a la Unidad de Cuidados Intensivos con tratamiento antibiótico (Cefotaxime 100 mg/Kg./d más Amikacina 15 mg/Kg./d). El día 16 ha perdido el 13.7% de su peso al nacer (peso: 3400g) persistiendo la hipotonía, con succión débil, regurgitaciones y vómitos. Hacia el día 19 se mantenía grave, hipoactivo, alimentándose por gabaje y con aumento de sólo 40 gramos de peso. El día 21 ha perdido 130 gramos más, tiene fiebre de 38°C, acidosis metabólica ligera. El día 23, se mantiene grave, en esta oportunidad se decidió agregar al tratamiento Dopamina e Hidrocortisona (25 mg/kg/día) con el objetivo de mejorar la respuesta sistémica a la Sepsis, habiendo recibido Intacglobín, plasma, glóbulos y enérgica antibioticoterapia. El día 25 ha mejorado y dos días después se retiró tratamiento dopaminérgico, el día 28 presenta recaída sin pérdida de peso, se detectó hemocultivo positivo a levaduras y la radiografía evolutiva mostró aumento de la trama pulmonar hiliobasal bilateral de aspecto inflamatorio, se renovó el tratamiento antibiótico endovenoso con Ceftriaxona (100 mg/kg/d) y Vancomicina (40mg/kg/d) y se mantuvo tratamiento con glucocorticoides. El día 30 ha perdido 180 gramos en las últimas 48 horas, comienza a sospecharse un "trastorno congénito del metabolismo". El día 31 tiene fiebre, está comprometido hemodinámicamente y se recibe hemocultivo positivo a Enterobacter, presenta fallo ventilatorio y convulsiones tónico-clónicas, por lo que se acopló a un ventilador, continúa perdiendo peso corporal y hacia el día 33 se interconsultó con Endocrinología planteándose una HSC clásica perdedora de sal; se tomó muestra de sangre para determinación de 17-OH-progesterona para confirmación del diagnóstico, y se sube la dosis de Hidrocortisona a 50 mg/Kg./día, se suspende la administración de diuréticos y potasio obteniéndose una respuesta rápida hacia la mejoría. ]]>
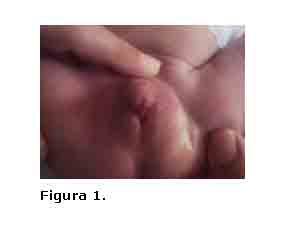
Caso 2.- B.L.B.E.Antecedentes prenatales: Madre primigesta con historia familiar negativa que ingresó a las 36 semanas por oligoamnios, no consanguinidad parental. Fue valorado en el Servicio de Neonatología por genitales externos ambiguos.
Se trata de recién nacido de aspecto saludable, 3180 gramos y 49 cm de talla con falo hipertrófico de aproximadamente 4,5 cm, pliegues labioescrotales rugosos, pigmentados, fusionados en sentido posteroanterior, con orificio miccional de 0.5 cm en la base del falo que recuerda la apertura de un seno urogenital, desde éste y hasta el extremo distal del falo se extienden dos cuerdas tendinosas. No se palpan gónadas en los pliegues labioescrotales (figura 1.). A los cinco días de edad se tomaron muestras de sangre para Cariotipo y determinación de 17-OH-progesterona, recomendándose observación estrecha ante la posibilidad de presentar signos de pérdida salina. Recibimos resultado del Cariotipo: 46, XX y cifra elevadas de 17-OH-progesterona (259 ng/ml), con lo que se corroboró el diagnóstico de HSC, iniciándose tratamiento con Fludrocortisona 0.1 mg/día y Acetato de Cortisona 25 mg/m²/día. No se presentaron desequilibrios hidroelectrolíticos.
DISCUSIÓN
La esteroidogénesis adrenal comprende las vías de síntesis a través de las cuales el colesterol se convierte en mineralocorticoides (Aldosterona), glucocorticoides (Cortisol) y andrógenos (Dehidroepiandrosterona), sistema controlado por el eje Hipotálamo- Hipófisis- Adrenal, siendo los niveles de Cortisol en sangre los encargados de estimular o inhibir este proceso a través de un mecanismo de retroalimentación.
Cuando la enzima deficitaria es la 21-hidroxilasa se compromete la síntesis de Cortisol, también puede estar interrumpida la producción de Aldosterona dando lugar a la sobreestimulación de la corteza suprarrenal por la ACTH con incremento del metabolito anterior al bloqueo metabólico (17-OH-progesterona) que deriva en aumento en la producción de andrógenos adrenales.1-7 La expresión clínica de este fallo metabólico está relacionada con:
Déficit de Cortisol, que produce mala respuesta de adaptación al stress e hipoglucemia
Déficit de Aldosterona, que ocasiona deshidratación con pérdida de sodio y retención de potasio.
Hiperproducción de Dehidroepiandrosterona, que determina la aparición de signos de androgenización (hipertrofia muscular, aparición de vello sexual, aceleración de la velocidad de crecimiento y de la maduración esquelética); en la hembra ocasionará virilización de los genitales externos. ]]>
La presencia en recién nacidos de genitales externos ambiguos, vómitos y desequilibrio hidoelectrolítico, sugiere el diagnóstico de HSC.1 En ambos casos se arribó a igual diagnóstico, sin embargo fue notable la diferencia en cuanto a la precocidad de éste, porque es fácil sospechar esta afección en las féminas por la característica virilización de genitales externos que muestran, lo que constituye una emergencia en la atención del recién nacido y obliga al manejo interdisciplinario para asignación de sexo legal. Ocurre todo lo contrario en el varón, que puede llegar al fallecimiento si no se establece el diagnóstico; nuestro caso varón presentó tempranamente manifestaciones clínicas de pérdida de sal (pérdida de peso, rechazo a los alimentos, vómitos y febrícula), síntomas y signos que pueden observarse en otros trastornos más frecuentes. Fue diagnosticado por exclusión después de múltiples interpretaciones diagnósticas. La respuesta al tratamiento específico para HSC de ambos pacientes fue satisfactoria.La HSC puede estar sujeta a muchas dificultades diagnósticas, ya que su cortejo sintomático es parecido al de otros errores congénitos del metabolismo, y no es el único trastorno capaz de producir ambigüedad genital.
Los casos presentados se diagnosticaron en Pinar del Río durante el año 2002, cuando aún no se disponía de pesquisaje neonatal para HSC por déficit de 21-hidroxilasa, que en estos momentos se realiza mediante la determinación de 17-OH-progesterona por inmunoensayo en muestra de sangre obtenida por punción del talón recogida en papel de filtro, en la primera semana de vida.
El establecer el diagnóstico de HSC es importante para el asesoramiento genético de la familia, ya que como trastorno autosómico recesivo se expresa en estado homocigótico, y ambos padres de un enfermo son heterocigóticos al gen mutado (portadores sanos), presentando como pareja un riesgo de recurrencia de un 25% en cada futuro embarazo.10
El diagnóstico prenatal de la deficiencia de 21-hidroxilasa durante el primer trimestre es factible,11-12 pero desafortunadamente no disponible aún en nuestro medio.
REFERENCIAS BIBLIOGRÁFICAS
1- Wilson T Congenital adrenal hyperplasia. Medicine.com, April 11.2003.
2-Oriola J. Diagnóstico molecular de los déficit de 21-hidroxilasa y su correlación con el fenotipo. Endocrinología y Nutrición 1999; 46: 168-72.
3-Carrol MC, Campbell RD, Porter RR Mapping of steroid 21-hydroxylase genes adjacent to complement components C4 genes in HLA, the mayor histocompatibility complex in man. Proc Natl Acad Sci USA 1985; 82: 521-525.
4-White PC, Grossberger D, Onufer BJ Two genes encoding steroid 21-hydroxylase are located near the genes encoding the fourth component of complement in man. Proc Natl Acad Sci USA 1985; 82: 1089-1093.
5-Alonso IT, Oriola J, Macía CB, De Sas MF, Rueda CC, Loidi L Hiperplasia adrenal congénita: correlación fenotípica-genotípica. A propósito de cinco casos. Endocrinología y Nutrición, Viernes 1 Diciembre 2000. Volumen 47 - Número 10 p. 290 - 293
6-Wedel A. Molecular approaches for the diagnosis of 21-hydroxylase deficiency and congenital adrenal hyperplasia. Clin Lab Med 1996 Mar:16(1):125-37.
7-White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocrin Rev 2000; 21: 245-91.
8-Pang S, Wallace MA, Hofman L, Thuline HC, Dorche C, Lyon ICT et al World-wide experience in newborn screenning for classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatrics 1988; 81: 866-874.
9-Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI High frequency of nonclassical steroid 21-hydroxilase deficiency. Am J Hum Genet,1985;37:650-667.
10-Guizar-Vazquez J(ed): Genética Clínica. Diagnóstico y manejo de las enfermedades hereditarias. Editorial Manual Moderno.Tercera Edición.México,2002.
11-Mornet E, Boue J, Raox-Demay M et al: First trimestre prenatal diagnosis of 21-hidroxilase deficiency by linkage analysis HLA-DNA probes and by 17-hydroxyprogesterone determination. Hum Genet 1986; 73:358-364.
12-Diukman R,Goldberg JD: Prenatal Diagnosis of Inherited Metabolic Diseases. En: Fetal Medicine (Special Issue) West J Med 1993; 159:374-381. ]]>
Recibido: 16 de Septiembre de 2006
Aprobado: 20 de Noviembre de 2006
Dr. José Rafael Hernández Gómez. Especialista de I Grado en Endocrinología. Asistente. Hospital Pediátrico "Pepe Portilla". Pinar del Río. Dirección: Pepe Portilla No. 71. Pinar del Río, Cuba ]]>