Reporte de familias con neurofibromatosis y otras enfermedades genéticas
Reports of families suffering from neurofibromatosis and other genetic disorders
Miladys Orraca Castillo1, Deysi Licourt Otero2, Ana Isabel Sánchez Álvarez de La Campa3 ]]>
1Especialista de Segundo Grado en Genética Clínica. Profesora Auxiliar. Centro Provincial de Genética Médica Pinar del Río. Correo electrónico: milgene@princesa.pri.sld.cu
2Especialista de Primer Grado en Medicina General Integral y Genética Clínica. Profesora Asistente. Centro Provincial de Genética Médica Pinar del Río. Correo electrónico: deysili@princesa.pri.sld.cu
3Especialista de Primer Grado en Medicina General Integral. Profesora Instructora. Sede universitaria Municipal San Juan y Martínez. Servicio Municipal de Genética Médica San Juan y Martínez. Correo electrónico: anai@princesa.pri.sld.cu
RESUMEN
La neurofibromatosis tipo 1, es una enfermedad genética que primariamente afecta el desarrollo y crecimiento celular del sistema nervioso, clínicamente se caracteriza por máculas café con leche, neurofibromas, pecas en regiones no expuestas al sol, nódulos de Lisch, lesiones óseas y glioma óptico. En el presente trabajo se describen dos familias, en las cuales algunos individuos padecen esta enfermedad y otros miembros de la misma familia muestran una diferente enfermedad genética. La coexistencia intrafamiliar de dos enfermedades genéticas diferentes es muy poco frecuente, es por ello, que se decide realizar la revisión y publicación científica sobre el tema.
DeCS: NEUROFIBROMATOSIS/genética/clasificación/diagnóstico, DISTROFIAS MUSCULARES/genética.
ABSTRACT
Neurofibromatosis type-1 is a genetic condition that affects the development and cellular growth of the nervous system, which is clinically characterized by multiple café-au-lait spots, neurofibromas, freckles in non-sun exposed regions, Lisch nodules, osseous lesions and optic glioma. The present paper describes two families, having some individuals suffering from this condition and other members of the same family who present a different genetic condition. The intrafamilial coexistence of two different genetic conditions is very rare, that is why a literature review and a scientific research about the topic were carried out.
DeCS: NEUROFIBROMATOSES/genetics/classification/diagnosis, MUSCULAR DISTROPHIES/genetics.
INTRODUCCIÓN
La neurofibromatosis tipo 1 (NF1) es un trastorno genético del sistema nervioso que causa el crecimiento de tumores en los nervios. Estos tumores son benignos, lo que significa que no son cancerígenos. Las NF1 también pueden provocar anomalías en la piel y los huesos, la gravedad de los síntomas varía enormemente. La enfermedad ocurre en todos los grupos étnicos y raciales del mundo y afecta a ambos sexos por igual. El gen de la NF1 se encuentra en el cromosoma 17. En aproximadamente el 50 % de los casos, el gen anormal se hereda del progenitor que tiene el trastorno. En algunos casos, el progenitor afectado puede tener síntomas leves y no ser consciente de que padece el trastorno. La otra mitad de los casos de NF1 son producto de nuevas mutaciones (cambios) en los genes causantes.1, 2
Las enfermedades de la piel con herencia autosómica dominante, en algunas ocasiones, presentan un fenotipo segmental; en recientes estudios moleculares se ha demostrado la existencia de un mosaicismo somático como manifestación clínica, se han delineado las manifestaciones segmentales de genodermatosis autosómicas dominantes, esta variante es caracterizada por una presentación clínica más difusa.3 ]]>
En el presente trabajo se realiza la descripción de dos familias en las que tienen algunos de sus miembros afectados con neurofibromatosis y además coexisten otras enfermedades genéticas diferentes, esto es un hallazgo no descrito en la literatura consultada, esta consideración constituye la principal motivación para la publicación y se ofrece para el conocimiento por parte de otros profesionales de la salud.MATERIAL Y MÉTODO
Se realizó un estudio descriptivo y transversal desde enero-diciembre del año 2010 en toda la provincia de Pinar del Río, a partir de 120 familias registradas con el diagnóstico de NF1, de las consultas del Centro Provincial de Genética de Pinar del Río. La muestra estuvo constituida por 15 pacientes, distribuidos entre dos familias sin lazos de consanguinidad, de ellos, solo cuatro presentan NF1 y el resto tienen otras enfermedades genéticas diferentes, en una familia solo dos personas integraban la muestra y el resto pertenecen a la otra familia.
El universo de pacientes se citó hacia el Centro Provincial de Genética de Pinar del Río y se visitaron en sus municipios para facilitar el trabajo y la selección de la muestra, a las 15 personas que integraban la muestra se le realizó la historia clínica genética previo consentimiento informado, confeccionó del árbol genealógico y fueron sometidos a un examen físico que estuvo conformado por varios aspectos:
Características de la piel (pigmentación, aumento de volumen, etc.)
Examen de regiones no expuestas al sol.
Características cráneo faciales.
Examen oftalmológico.
Examen neurológico.
Mensuraciones. ]]>
RESULTADOS
Presentación de dos familias: familia 1, perteneciente al Municipio San Juan y Martínez, Provincia Pinar del Río, representada por cinco miembros, tres de ellos, del sexo masculino y dos del sexo femenino que acuden a los servicios de genética por presentar alteraciones dérmicas, en dos de sus integrantes. Figura 1
Paciente II-2, propósito afectado con NF1, 18 años de edad. Datos positivos al examen físico:
Piel: más de seis manchas café con leche que se distribuyen en hemitórax izquierdo, en la región anterior y posterior del cuello y el tronco con diámetros variables que oscilan desde 0,5 hasta 5 cms, con bordes bien definidos. Se presentan además pecas en región axilar izquierda con diámetros de 0,1-0,3 cms, incontables en cuanto al número de ellas.
Exámenes Complementarios:
Rx de cráneo: Displasia y esclerosis del ala mayor del esfenoides, esclerosis de las órbitas y aumento del diámetro anteroposterior de la silla turca. Cariotipo: 46, XY.
Coeficiente de inteligencia: percentil 10 Rango IV. Inferior al técnico medio.
Examen oftalmológico: en el ojo izquierdo más de 10 nódulos de Lisch en el sector inferior temporal. Ojo derecho, siete nódulos de Lisch en el sector inferior nasal y temporal.
Paciente II-3, de 11 años de edad. Datos positivos al examen físico:Miembro inferior derecho: Aumento de volumen con presencia de hemangiomas. ]]>
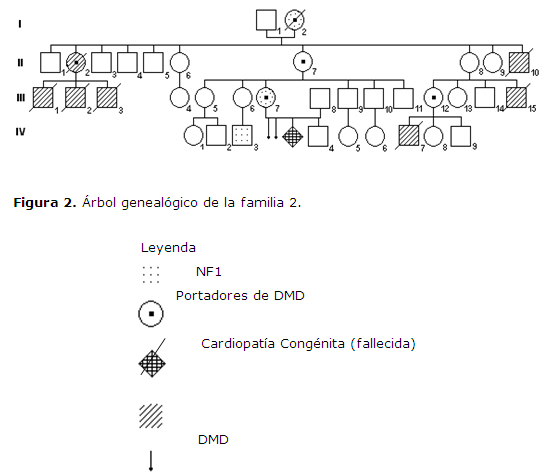
Familia 2, perteneciente al Municipio Sandino, Provincia Pinar del Río, representada por 36 miembros, 20 de ellos del sexo masculino y los que restan del sexo femenino, el propósito III-7 acude a los servicios de genética por sospecha de embarazo, y antecedentes familiares de individuos con distrofia muscular de Duchenne y otros miembros de la familia con manchas café con leche. Figura 2
Paciente III-7, caso propósito, de 32 años de edad. Datos positivos al examen físico: piel con mancha café con leche que se distribuye en la región anterior y derecha del cuello y el tronco extendiéndose hasta la parte proximal del brazo derecho. Se presentan además pecas en región axilar izquierda con diámetros de 0,1-0,3 cms, incontables en cuanto al número de ellas.
Exámenes complementarios: examen oftalmológico: en el ojo izquierdo se observan tres nódulos de Lisch. Estudio de portadoras por análisis de ligamiento a varias mujeres de la familia. Resultaron portadoras del gen Duchenne: I-2, II-2, II-7, II- 12, III-7.
Se trata de una paciente con embarazo en la que se concluye diagnóstico definitivo de: neurofibromatosis segmentaria o tipo 5 y portadora para la Distrofia Muscular de Duchenne.
DISCUSIÓN
La NF1 es una enfermedad altamente variable en su expresión, con síntomas y signos que pueden comenzar al nacimiento y continuar para toda la vida, los hallazgos fenotípicos pueden ser divididos en manifestaciones tumorales y no tumorales; estas últimas incluyen manifestaciones pigmentarias, displasias esqueléticas y vasculares así como discapacidad intelectual, como se describe en el propósito de la familia 1, con excepción de las complicaciones vasculares que no están presentes en este caso, pero es interesante que la hermana del propósito padezca de una enfermedad genética diferente, llamada Klippel-Trenaunay-Weber, es un síndrome con hemangiomas e hipertrofia de los tejidos y puede acompañarse de severos defectos cardiacos y en la misma se reporta agregación familiar lo cual no se presenta en esta familia, no se encuentra reportes en la literatura de la coexistencia de dos enfermedades genéticas diferentes en una misma familia.4, 5,6
La NF1 es una enfermedad progresiva, de manera que las diferentes manifestaciones y complicaciones van apareciendo con el decursar de los años. Los criterios diagnósticos fueron establecidos en 1988 y son necesarios al menos 2 de ellos para determinar una neurofibromatosis.7, 8
La neurofibromatosis sectorial o segmental, que recibe la clasificación de neurofibromatosis tipo 5 o NF1 localizada en mosaico, está presente en el propósito de la familia 2, representa un mosaicismo somático; se produce por una microdeleción de origen postcigótico en el gen NF1 que es observada solo en los tejidos que proceden de las manchas café con leche o de otras alteraciones dérmicas propias de la enfermedad , y es ausente en fibroblastos que proceden de la piel normal así como también en los leucocitos de sangre periférica, la frecuencia relativa con la que se presenta es de un 0.0018%, siendo alrededor de 30 veces menos frecuente que la neurofibromatosis tipo 1.9
El área involucrada en la NF segmental no solo se limita a áreas pequeñas en el cuerpo o a un cuadrante sino que también incluye segmentos del cuerpo a ambos lados de la línea media de forma simétrica o asimétrica, en el área afectada puede haber manchas café con leche con o sin pecas, como se describe en la paciente propósito de la familia 2, o neurofibromas o una combinación de las anteriores, se puede encontrar en estas zonas malignización de las lesiones como complicación de la enfermedad pero la prevalencia para la presencia de este hallazgo es menor del 5%. ]]>
Algunos autores han clasificado la neurofibromatosis segmental en cuatro grupos: pacientes con cambios pigmentarios solamente, pacientes con neurofibromas, los pacientes que tienen una combinación de los dos anteriores, y aquellos que solo presentan neurofibromas plexiformes. La significación clínica de estos grupos es por la edad de presentación de estos signos clínicos.10La paciente propósito de la familia 2, además de presentar una NF segmental, es portadora de una enfermedad genética neurodegenerativa llamada distrofia muscular de Duchenne y tiene antecedentes de una interrupción de embarazo por padecer el feto de una cardiopatía congénita compleja, lo anterior pudiera ser una manifestación severa prenatal de la neurofibromatosis, pero no se reporta en la literatura este hallazgo, ni tampoco la segregación en una misma familia de varias enfermedades genéticas.
Los hallazgos encontrados en ambas familias son novedosos y de gran interés para los profesionales de la salud y sobre todo los que trabajan en la disciplina de la genética médica y que brindan el asesoramiento genético a pacientes y familias en riesgo o afectadas por alguna enfermedad genética.
REFERENCIAS BIBLIOGRÁFICAS
1. Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1. Pediatrics. [Internet]. 2009 [Citado mayo 2011]; 123(1): [Aprox. 10p.]. Disponible en: http://www.pediatrics.org/cgi/content/full/123/1/124
2. Haggstrom A, Frieden I. Segmental Hemangioma: An Important Clinical Term. American Journal of Medical Genetics Part A. [Internet]. 2008 [Citado mayo 2011]; 146(5): [Aprox. 1p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/18241074
3. Theos A, Bruce R. Pathophysiology of Neurofibromatosis Type 1. Annals of Internal Medicine. [Internet]. 2006 [Citado mayo 2011]; 144(11): [Aprox. 7p.]. Disponible en:http://www.ncbi.nlm.nih.gov/pubmed/16754926
4. Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. [Internet]. 2007 [Citado mayo 2011]; 44(2): [Aprox. 7p.]. Disponible en: http://jmg.bmj.com/content/44/2/81.full
5. K Janniger C . Klippel-Trenaunay-Weber Síndrome. [Internet]. Medscape; 2010 [Citado Mayo 2011]. Disponible en: http://emedicine.medscape.com/article/1084257-overview
6. Beiro AC, Carvalho Dantas JF, Conte Neto N, Bastos Nasciben M, Scarso Filho J. Neurofibromatose: uma desordem hereditária: relato de caso de ocorrência em mãe e filha. Rev Ciênc Méd Biol. [Internet]. 2008 [Citado mayo 2011]; 7(2): [Aprox. 4p.]. Disponible en: http://www.portalseer.ufba.br/index.php/cmbio/article/view/4453
7. Gonçalves KS, Geller M, Soares de Mouro NR, Lopes SV. Genética da neurofibromatose tipo 1. Rev Ciênc Méd Biol. [Internet]. 2007 [Citado mayo 2011]; 6(3): [Aprox. 10p.]. Disponible en: http://www.portalseer.ufba.br/index.php/cmbio/article/view/4396/3224
8. Ruggieri M. Mosaic (Segmental) Neurofibromatosis Type 1 (NF1) and Type 2 (NF2): No Longer Neurofibromatosis Type 5(NF5). American Journal of Medical Genetics. [Internet]. 2001 [Citado mayo 2011]; 101(2): [Aprox. 2p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/11391665
Recibido: 2 de septiembre de 2011.
Aprobado: 15 de noviembre de 2011.
]]> Dra. Miladys Orraca Castillo. Especialista de Segundo Grado en Genética Clínica. Profesora Auxiliar. Centro Provincial de Genética Médica Pinar del Río. Correo electrónico: milgene@princesa.pri.sld.cu ]]>
{kind=link}
{kind=link}