{kind=link}

Estudio clínico-genético de pacientes cubanos con síndrome de West
Clinical and genetic studies in Cuban patients suffering from West syndrome
Anitery Travieso Tellez1, Araceli Lantigua Cruz2, Ramiro García García3
1Especialista de Primer Grado en Medicina General Integral y Genética Clínica. Centro Provincial de Genética Médica. Pinar del Río. Correo electrónico: any0511@princesa.pri.sld.cu ]]>
2Especialista de Segundo Grado en Genética Clínica. Profesora Titular. Centro Nacional de Genética Médica. La Habana. Correo electrónico: lantigua@infomed.sld.cu
3Especialista de Primer Grado en Neurología y de Segundo Grado en Pediatría. Profesor Titular. Hospital Pediátrico Juan Manuel Márquez. La Habana. Correo electrónico: ramirogg@infomed.sld.cu
RESUMEN
El síndrome de West constituye una encefalopatía epiléptica asociada a una amplia diversidad de factores causales, cuyas bases genéticas hasta el momento no se han estudiado en Cuba. Con el objetivo de describir las principales características clínicas y genéticas del trastorno, se realizó un estudio descriptivo, transversal en los pacientes con el síndrome de West, atendido en dos hospitales pediátricos de La Habana desde enero de 2005 a diciembre del año 2009. Predominaron los pacientes masculinos. Inicialmente los casos sintomáticos representaron solo el 53.85% del total. Se identificó la historia familiar positiva de epilepsia en el 51.92% y hubo una recurrencia del diagnóstico de síndrome West en tres familias; lo que orienta hacia la participación del componente genético en el desarrollo de este síndrome epiléptico. El 82.69% presentó hallazgos positivos al examen físico, de ellos, el 11.63% mostró anomalías cromosómicas. Las pruebas metabólicas aportaron diagnóstico en el 17.39% de los casos con antecedentes familiares positivos y/o defectos congénitos. Finalmente se modificó la frecuencia de clasificación del síndrome epiléptico concluyendo el estudio con un 78.85% de casos sintomáticos lo que apoya la utilidad del enfoque genético en la evaluación de los pacientes con síndrome de West.
DeCS: ESPASMOS INFANTILES/clasificación/etiología/genética .
ABSTRACT
West Syndrome, which is classified as an epileptic encephalopathy, is associated with an ample variety of etiological factors. Up to this moment, the molecular bases of this entity have not been studied in Cuba. A cross-sectional, descriptive study of West Syndrome was conducted with the purpose of describing the main clinical and genetic characteristics of this syndrome. It included the patients diagnosed in two pediatric hospitals in Havana between January 2005 and December 2009. The study showed a high prevalence of male patients. Initially, symptomatic cases represented only 53.85 % of the sample. A positive family history of epilepsy was detected in 51.92 % of the cases and recurrence of the disease was identified in three of all families included in the study. These two results pointed to a strong genetic component in association with the development of West Syndrome. The 82.69% was found to have positive physical examination findings; among them 11.63 % presented chromosomal anomalies. Metabolic studies confirmed 17.39% of the cases with family history and/or congenital defects. At the end, the frequency of classification for the epileptic syndrome was modified to conclude the study with a 78.85 % out of the symptomatic cases. Results corroborate the importance of a genetic assessment in the evaluation and diagnosis of patients with suspected West Syndrome.
DeCS: INFANTILE SPASMS/classification/etiology/genetics.
INTRODUCCIÓN
La contribución de la genética en el diagnóstico y seguimiento de las epilepsias y los síndromes epilépticos no se discute en la actualidad. Los servicios de referencia en Neurología a nivel mundial incluyen la evaluación y los estudios genéticos dentro del organigrama habitual para el diagnóstico etiológico y el manejo de los pacientes con epilepsia, tenga esta una causa desconocida o "aparentemente identificada".1
Una de las formas de presentación de la epilepsia de comienzo en la infancia temprana, cuya aparición se relaciona con la edad y que en sí misma no puede considerarse una entidad nosológica es el síndrome de West; constituye un heterogéneo grupo de condiciones que comparten la triada clínica de espasmos epilépticos, deterioro del neurodesarrollo y patrón de hipsarritmia en el electroencefalograma (EEG).2
La discapacidad cognitiva que se presenta en el 90% de los casos afectados puede ser de diversos grados y con frecuencia se asocia también a déficit motor, a trastornos de conducta y rasgos autísticos. Justamente, uno de sus aspectos clínicos más sobresalientes se centra en la magnitud de las consecuencias que produce en el orden del neurodesarrollo este tipo peculiar de síndrome epiléptico.3
El pronóstico, aunque generalmente grave, resulta incierto dada las peculiaridades evolutivas de esta entidad y debido a la multiplicidad de sus bases patológicas.2
En Cuba, pese a los avances científicos alcanzados en el campo de la Neurología, aún se investiga poco sobre las bases genéticas de la epilepsia, situación que compromete el diagnóstico etiológico de síndrome de West, y por tanto, el manejo individualizado de estos pacientes. Ante dicha realidad, la presente investigación asume como propósito describir las características clínico-genéticas del síndrome de West. Es el primer estudio de carácter descriptivo en el país aplicado al tema de las encefalopatías epilépticas desde la perspectiva genética.
Disponer de una estrategia de evaluación genética para la investigación etiológica de estos pacientes representa una mayor probabilidad de clasificar cada caso según su origen, de forma precoz y acertada. La prontitud de los resultados contribuye a los tratamientos más rápidos y específicos en la medida de las posibilidades, lo que redunda en la calidad de vida de los afectados y reduce los costos sociales por criterio de discapacidad. Orienta además hacia pronósticos mejor fundamentados.4
]]> MATERIAL Y MÉTODO
Se realizó un estudio observacional, descriptivo, de corte transversal en una muestra de 52 pacientes con diagnóstico clínico y electroencefalográfico de síndrome de West, atendidos en los hospitales pediátricos Juan Manuel Márquez y William Soler de La Habana desde enero de 2005 a diciembre de 2009. Se consideraron como criterios de inclusión la edad de los pacientes menor de 6 años y el consentimiento de padres o tutores para incluirse en el estudio. Fueron excluidos los niños que fallecieron durante el transcurso de la investigación por razones no relacionadas directamente con el síndrome epiléptico, los pacientes sin resultados de pruebas imagenológicas, metabólicas y cromosómicas y aquellos inasistentes por cualquier causa a las evaluaciones programadas.
Los pacientes fueron citados para evaluación en la consulta de genética clínica, previa revisión de sus respectivas historias clínicas hospitalarias. Se realizó la entrevista a los familiares, indagando sobre antecedentes familiares de epilepsia y se procedió al examen físico de los casos.
Fueron indicados los estudios de neuroimagen a todos los participantes en la investigación. Los niños con antecedentes familiares de epilepsia y/o aquellos con hallazgos positivos al examen físico dismorfológico, se sometieron además a estudios metabólicos y cromosómicos. Los datos disponibles de cada paciente se plasmaron en un modelo de recogida de información resumiendo de manera individualizada el comportamiento de las variables de interés en nuestro estudio. Al modelo se adjuntó el árbol genealógico de cada paciente. Una vez concluido el estudio, con los datos de cada paciente incluyendo los resultados de las investigaciones de laboratorio se evaluó nuevamente la clasificación de cada uno según la etiología del síndrome epiléptico. Se aplicaron los criterios de la Liga Internacional contra la epilepsia en el 2001:
- Síndrome de epilepsia sintomática: La causa de la epilepsia ha sido comprobada (tumor, lesión vascular, etc.) De manera habitual no evoluciona con respuesta satisfactoria a la medicación, puede tener afectación intelectual y/o motora, el resultado del EEG es anormal.
- Síndrome de epilepsia probablemente sintomática: Empleado para definir los síndromes que se presumen sean sintomáticos, pero cuyo origen no ha sido identificado. La evolución es similar a la de las epilepsias sintomáticas, o sea, no hay respuesta a la medicación, afectación intelectual y/o motora y EEG anormal.
Los resultados del análisis de las variables cualitativas se resumieron en frecuencias absolutas, razones y por cientos. Para por ciento de positividad del cariotipo a los pacientes a los que se les realizó y para la presencia de defectos congénitos en la totalidad de pacientes, se determinó el intervalo de confianza (IC), con 95% de confiabilidad.
RESULTADOS
En el estudio se observó un predominio del sexo masculino. La razón de masculinidad fue de 1.74. Se clasificaron los pacientes según la etiología del trastorno epiléptico. Se obtuvo un ligero predominio de los casos sintomáticos, es decir, aquellos en los que se había identificado un factor causal; representando más de la mitad del total. De ellos, la mayor proporción estuvo dada por el sexo masculino. Este predominio se repitió en el grupo de casos probablemente sintomáticos, donde la diferencia entre sexos fue aun más notable. Los varones constituyeron el doble del número de pacientes femeninas en este subgrupo, tabla 1. ]]>
Al indagar sobre la historia familiar de epilepsia entre los pacientes con síndrome de West, se obtuvo que más de la mitad presentó antecedentes familiares positivos (27/52), predominando ligeramente en el grupo de los casos probablemente sintomáticos. Los parientes de segundo grado resultaron ser los mayormente afectados, específicamente los tíos y abuelos, tabla 2.Cuando se exploró sobre antecedentes familiares de síndrome de West específicamente, se encontraron que 4 pacientes pertenecientes a tres familias, tienen algún pariente con diagnóstico clínico y electroencefalográfico de este mismo trastorno. Dos de los pacientes pertenecen a la misma familia, pues son hermanos y ambos cumplen con los criterios de inclusión en el estudio, por lo que en este acápite se manejan como un solo caso.
Es decir, que en la investigación se estudiaron 52 pacientes en 51 familias. De estas, 3 presentaron recurrencia del síndrome de West. Los familiares concordantes para este diagnóstico presentaron características semejantes entre ellos en cuanto a edad de comienzo de los síntomas, caracterización de los espasmos, clasificación etiológica y evolución de la encefalopatía, menos en una familia en que siendo ambos parientes diagnosticados con síndrome de West de tipo sintomático, los eventos presumiblemente desencadenantes son totalmente diferentes. De las tres familias, en dos de ellas existe un tercer familiar afecto con otro tipo de epilepsia.
Según la presencia o no de defectos congénitos, predominaron los pacientes con positividad para este criterio, en relación al total de la muestra, 43/52 lo que representa el 82.69% (IC; 69.7-91.8). De ellos, la mayoría estuvo representada por casos con defectos menores y 12 pacientes solo 1 defecto menor, mientras que 15 presentaron entre 1y 3 defectos menores y en igual número de casos se identificaron más de 3 defectos. Los pacientes con defectos mayores resultaron todos casos sintomáticos, y de ellos, 3 tuvieron asociadas más de tres anomalías menores, tabla 3.
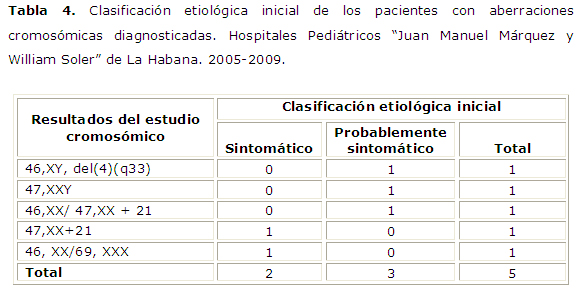
Del total de pacientes, solo con la exploración física se logró identificar la causa del trastorno en dos casos, clasificados hasta el momento del estudio como probablemente sintomáticos y que resultaron ser dos síndromes neurocutáneos: Un caso de neurofibromatosis tipo1 (NF1) y un caso de Hipomelanosis ito. En el estudio cromosómico realizado se encontraron 5/43 pacientes con diagnóstico positivo, que representaron el 11.63% del total de casos (IC; 3.87-25.1) a los que se realizó el cariotipo. Se muestra la clasificación etiopatogénica inicial de dichos pacientes, tabla 4.
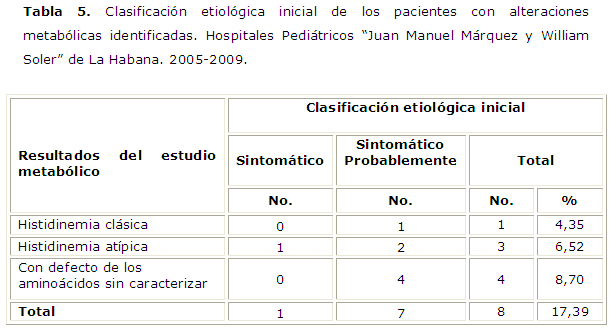
Se realizó un estudio metabólico a un total de 46 pacientes, de los que 24 compartieron antecedentes patológicos familiares de epilepsia y la presencia de defectos congénitos, 19 solo tuvieron defectos congénitos y 3 solo el antecedente familiar positivo. Todos tuvieron defectos a nivel de los aminoácidos, en 4 casos, logró caracterizarse el defecto, y en el resto, quedó planteado el diagnóstico de un error innato del metabolismo pendiente a ser definido, tabla 5.
Luego de concluido el estudio el número de pacientes con síndrome West sintomático aumentó comparativamente con el inicio. En 13 pacientes anteriormente clasificados como probablemente sintomáticos, fue identificada la etiología del síndrome epiléptico. Se concluyó con un total de 41 pacientes sintomáticos y solo 11 permanecieron sin causa identificable.
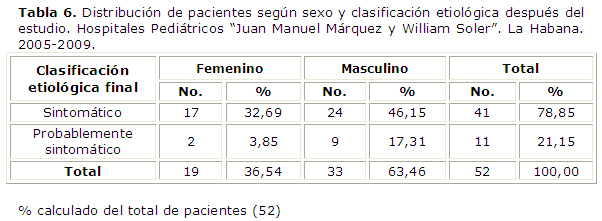
Se realizó nuevamente la distribución de pacientes según el sexo y la clasificación etiológica después del estudio, resultados que se muestran en la tabla 6 . En el grupo de casos sintomáticos la razón de masculinidad (1.41) disminuyó ligeramente con relación a la calculada al inicio del estudio (1.74). Sin embargo, el predominio de los varones en el grupo probablemente sintomático es notable. La razón de masculinidad aumentó a 4.5, es decir que al concluir el estudio hubo casi 5 varones con síndrome de West probablemente sintomático por cada paciente femenina incluida en el grupo.
]]> DISCUSIÓN
El predominio de varones en el presente estudio, con una proporción aproximada 2:1 con relación a las hembras, coincide con los reportes de las investigaciones relacionadas sobre el tema que llegan a tener indicadores de hasta 3 varones por cada hembra.5,6
Se ha expuesto que el cromosoma X contiene numerosos loci relacionados potencialmente con el desarrollo del Sistema Nervioso Central. De hecho, en este cromosoma parecen existir proporcionalmente más genes asociados con la cognición que en los autosomas. El estado hemicigótico de los varones para los genes ubicados en el cromosoma X constituye una desigualdad biológica que los coloca en un riesgo superior con relación a las hembras para un número mayor de problemas de salud, que no necesariamente tienen que responder a síndromes monogénicos producidos por mutaciones. Cuando existen alteraciones de la metilación que afectan uno o más genes del cromosoma X del varón, estos son silenciados o sobre expresados parcialmente, lo que predispone a alteraciones del neurodesarrollo.7,8
Dentro de los genes identificados que codifican para factores de transcripción, localizados a nivel del cromosoma X se encuentra justamente el ARX, cuyas mutaciones son responsables hasta el momento del 5% aproximadamente de los casos de varones con fenotipo de espasmos infantiles; al tiempo que se plantea su posible acción como modificador de la expresión de otros genes que se expresan en cerebro y participan en los procesos de maduración del mismo. Ello confirma la justificación del predominio del síndrome de West en los varones y sugiere que no solo las mutaciones a nivel de este gen pueden ser causales del trastorno sino el efecto de mecanismos epigenéticos que modifiquen su expresión, alterando consecutivamente la función de otros genes que se encuentren bajo el efecto de ARX.9, 10
Por su parte, el porcentaje total de casos sintomáticos de acuerdo a los resultados obtenidos se sitúa por debajo de los últimos reportes que muestran una frecuencia de casos sintomáticos entre el 60% y el 70% del total de afectados con síndrome de West.2 Estos datos sugieren la necesidad de un abordaje más completo del paciente con espasmos infantiles en nuestro medio, a fin de lograr cada vez mejores y más rápidos procesos diagnósticos en función de la etiología del síndrome epiléptico.
La presencia de historia familiar positiva según los antecedentes de epilepsia en otros miembros, coincide con reportes de la literatura que orientan hacia la existencia de elementos genéticos que suponen un genotipo predispuesto a la posible acción de factores ambientales dando lugar al desarrollo de formas de epilepsia, como respuesta a alteraciones de la actividad neuronal dentro de la misma genealogía con un amplio espectro de variabilidad clínica.11
Este supuesto es apoyado por la recurrencia del diagnóstico de síndrome de West en tres familias, donde los familiares concordantes presentaron entre ellos las mismas características clínicas, la misma evolución y clasificación etiopatogénica, datos que coinciden con los resultados de un estudio similar en Japón12 que logró demostrar la agregación familiar para este síndrome por medio de un diseño de casos y controles. Durante la exploración física de los pacientes incluidos en la muestra se constató el valor insustituible del examen físico minucioso en los trastornos del neurodesarrollo, pues los hallazgos sugieren las posibles causas subyacentes, sobre todo las de origen genético de comienzo prenatal que dejan su huella física de acuerdo al momento del desarrollo en que produjeron las alteraciones.1
Se comprobó como la presencia de más de tres defectos congénitos menores se asocia generalmente con defectos congénitos mayores. Los síndromes neurocutáneos diagnosticados clínicamente se reportan entre las asociaciones etiológicas del síndrome de West, y no requieren de estudios complementarios para su confirmación si se obtienen clínicamente los criterios requeridos para su diagnóstico. En estos casos, la exploración imagenológica contribuye a la identificación de lesiones cerebrales compatibles con el diagnóstico del síndrome neuroectodérmico.13, 14
Por su parte, el estudio cromosómico aportó un diagnóstico etiológico en tres casos, y en otro paciente ofreció la explicación a los hallazgos detectados por exploración imagenológica, concluyéndose así el diagnóstico y facilitando un adecuado proceso de asesoramiento genético para la familia. Todos presentaron al examen físico algún defecto congénito, ya fuera facial, genital o en las extremidades, así como deterioro del neurodesarrollo. Estos hallazgos se corresponden con lo ampliamente reportado en la bibliografía sobre la asociación del síndrome West con diferentes cromosomopatías.2, 3,4
Los resultados obtenidos en los estudios cromosómicos, apoyan la propuesta del grupo de trabajo para los espasmos infantiles de la Liga Internacional contra la Epilepsia (ILAE) que en su más reciente reporte del 2010, sugiere la realización de estudio cromosómico a todo niño con espasmos infantiles y dismorfias, que luego de realizada la historia individual, el examen físico y las exploraciones imagenológicas se mantenga sin causa definida para su trastorno.4 ]]>
Actualmente no se niega la asociación de síndrome West y los errores innatos del metabolismo, apareciendo implicados una amplia variedad de estos.2,3 Campistol J en su artículo del 2003 en la Revista de Neurología muestra una lista de los defectos en el metabolismo de los aminoácidos asociados a síndrome de West. Entre ellos, cita la fenilcetonuria, la histidinemia, la tirosinemia III, la hiperprolinemia tipo I, la hiperglicinemia y el déficit de serina.15Sus observaciones coinciden con el resultado de los cuatro pacientes que en el estudio no pudieron ser caracterizados pero que mostraron un aumento de las concentraciones de determinados aminoácidos en suero, lo cual sugiere ampliar las investigaciones en estos pacientes, a fin de identificar, hasta donde nos lo permita la tecnología disponible, el defecto concreto que padecen. Finalmente, los resultados de la evaluación genética aportan nuevos casos sintomáticos al total de la muestra.
Ello remite nuevamente a las sugerencias de protocolos de estudio para espasmos infantiles que incluyen, además del habitual interrogatorio, el examen físico neurológico y exploración imagenológica; el enfoque dismorfológico y los estudios metabólicos y cromosómicos en aquellos casos que luego de los procederes habituales no han sido aún identificados según su etiología. Se sugiere, además, los estudios microbiológicos para las infecciones TORCHS en los casos que el examen físico así lo sugiera16, y por último, el estudio molecular en busca de las mutaciones más comúnmente asociadas a la aparición de síndrome de West.4
Cuando cada paciente se maneja de forma integral y según estos protocolos de estudio, las causas responsables del trastorno se definen progresivamente y son cada vez menos los pacientes probablemente sintomáticos.
Resulta llamativo que del grupo de 11 pacientes que al final del estudio quedaron sin causa identificada, el mayor por ciento esté representado por el sexo masculino. Podría inferirse que se trata de casos con espasmos infantiles ligados al X, genotípicamente predispuestos para esta encefalopatía epiléptica. La confirmación de esta hipótesis enfrenta a un nuevo problema con vistas a estudios futuros.
REFERENCIAS BIBLIOGRÁFICAS
1. Pal DK, Pong AW, Chung WK. Genetic evaluation and counseling for epilepsy. Nat Rev Neurol. [Internet]. 2010 [Citado 20 de mayo de 2011]; 6(8): [Aprox. 8p.]. Disponible en: http://www.nature.com/nrneurol/journal/v6/n8/full/nrneurol.2010.92.html
2. Fois A. Infantile spasms: review of the literature and personal experience. Ital J Pediatr. [Internet]. 2010 [Citado 20 de mayo de 2011]; 36: [Aprox. 1p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2829573/?tool=pubmed
3. Shields WD. Infantile Spasms: Little Seizures, BIG Consequences. Epilepsy Curr. [Internet]. 2006 [Citado 20 de mayo de 2011]; 6(3): [Aprox. 6p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1464162/?tool=pubmed
4. Pellock MJ, Hrachovy R, Shinnar S, Baram TZ, Bettis D, Dlugos DJ, et al. Infantile spasms: A U.S. consensus report. Epilepsia. [Internet]. 2010 [Citado 20 de mayo de 2011]; 51(10): [Aprox. 14p.]. Disponible en: http://onlinelibrary.wiley.com/doi/10.1111/j.1528-1167.2010.02657.x/full
5. Auvin S, Lamblin MD , Pandit F , Vallée L , Bouvet-Mourcia A . Infantile epileptic encephalopathy with late-onset spasms: report of 19 patients. Epilepsia. [Internet]. 2010 [Citado 20 de mayo de 2011]; 51(7): [Aprox. 6p.]. Disponible en: http://onlinelibrary.wiley.com/doi/10.1111/j.1528-1167.2010.02534.x/full
6. Hino-Fukuyo N, Haginoya K, Iinuma K. Epidemiological and clinical studies of West syndrome in Miyagi Prefecture, Japan. No To Hattatsu. [Internet]. 2007 [Citado 20 de mayo de 2011]; 39(4): [Aprox. 4p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/17633081
7. Nguyen DK, Disteche CM. High expression of the mammalian X chromosome in brain. Brain Res. [Internet]. 2006 [Citado 20 de mayo de 2011]; 1126(1): [Aprox. 3p.]. Disponible en: http://www.sciencedirect.com/science/article/pii/S0006899306024565
8. Jones JR, Skinner C, Friez MJ, Schwartz Ch, Stevenson RE. Hypothesis: Dysregulation of methylation of brain-expressed genes on the X chromosome and autism spectrum disorders. Am J Med Genet. [Internet]. 2008 [Citado 20 de mayo de 2011]; 146 (17): [Aprox. 7p.]. Disponible en: http://www.utdallas.edu/~mxa049000/lessons/research/literature/Autism/new/Genemethylandautismrev%2009.pdf
9. Kitamura K, Itou Y, Yanazawa M, Ohsawa M, Suzuki-Migishima R, Umeki Y, et al. Three human ARX mutations cause the lissencephaly-like and mental retardation with epilepsy-like pleiotropic phenotypes in mice. Hum Mol Genet. [Internet]. 2009 [Citado 20 de mayo de 2011]; 18(19): [Aprox. 16p.]. Disponible en: http://www.mendeley.com/research/three-human-arx-mutations-cause-lissencephalylike-mental-retardation-epilepsylike-pleiotropic-phenotypes-mice/
10. Friocourt G, Parnavelas JG. Mutations in ARX Result in Several Defects Involving GABAergic Neurons. Front Cell Neurosci. [Internet]. 2010 [Citado 20 de mayo de 2011]; 4. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2841486/
11. Dulac O, Feingold J, Plouin P, Chiron C, Pajot N, Ponsot G. Genetic predisposition to West syndrome. Epilepsia. [Internet]. 1993 [Citado 20 de mayo de 2011]; 34(4): [Aprox. 5p.]. Disponible en: http://www.ilae.org/visitors/centre/ctf/west_syndrome.cfm
12. Sugai K, Fukuyama Y, Yasuda K, Fujimoto S, Ohtsu M, Ohta H, et al. Clinical and pedigree study on familial cases of West syndrome in Japan. Brain & development. [Internet]. 2001 [Citado 20 de mayo de 2011];23(7):[Aprox. 6p.]. Disponible en: http://www.sciencedirect.com/science/article/pii/S0387760401002625
13. Arce Portillo E, Rufo-Campos M, Muñoz-Cabello B, Blanco-Martínez B, Madruga-Garrido M, Ruiz-Del Portal L, et al. Síndrome de West: etiología, opciones terapéuticas, evolución clínica y factores pronósticos. Rev Neurol. [Internet]. 2011 [Citado 20 de mayo de 2011]; 52: [Aprox.8p.]. Disponible en: http://www.neurologia.com/pdf/Web/5202/bf020081.pdf
14. Chandra PS, Salamon N, Nguyen ST. Infantile spasm-associated microencephaly in tuberous sclerosis complex and cortical dysplasia. Neurology. [Internet]. 2007 [Citado 20 de mayo de 2011]; 68(6): [Aprox. 7p.]. Disponible en: http://www.neurology.org/content/68/6/438.long
15. Campistol J, García-Cazorla A. Síndrome de West. Análisis, factores etiológicos y opciones terapéuticas. Rev Neurol. [Internet]. 2003 [Citado 20 de mayo de 2011]; 37(4): [Aprox. 7p.]. Disponible en: http://www.neurologia.com/pdf/Web/3704/p040345.pdf
16. Vigevano F, Bartuli A. Infantile epileptic syndromes and metabolic etiologies. J Child Neurol. [Internet]. 2002 [Citado 20 de mayo de 2011]; 17 (Suppl 3): [Aprox. 4p.]. Disponible en: http://web.ebscohost.com/ehost/pdfviewer/pdfviewer?sid=352915bf-e57f-449d-8d58-170ef619d185%40sessionmgr11&vid=2&hid=125
Recibido: 29 de noviembre de 2011.
Aprobado: 4 de mayo de 2011.
Dra. Anitery Travieso Tellez. Especialista de Primer Grado en Medicina General Integral y Genética Clínica. Centro Provincial de Genética Médica. Pinar del Río. Correo electrónico: any0511@princesa.pri.sld.cu ]]>
{kind=link}
{kind=link}
{kind=link}