

Síndrome de Saethre-Chotzen
Saethre-Chotzen syndrome
Elayne Esther Santana Hernández 1, Rafael Alfredo LLauradó Robles 2
]]> 1 Médica. Especialista de Segundo Grado en Medicina General Integral y Genética Clínica. Máster en Atención Integral al Niño Profesor Auxiliar. Investigador Agregado. Centro Provincial de Genética Médica de Holguín. Cuba. elsantana@infomed.sld.cu2 Médico.Especialista de Segundo Grado en Pediatría. Profesor Consultante. Investigador Agregado. Hospital Pediátrico Provincial Octavio de la Concepción de la Pedraja. Holguín. Cuba. rllaurado@infomed.sld.cu
Recibido: 17 de enero de 2017
Aprobado: 28 de abril de 2017
RESUMEN
El síndrome Saethre-Chotzen se encuentra entre las craneosinostosis hereditaria que se caracteriza por una sinostosis, uni o bilateral, precoz, de las suturas coronales, asimetría facial, ptosis y en ocasiones estrabismo. Se presenta una paciente femenina de 10 años con craneosisnostosis que le produjo asimetría de cráneo plagiocefacia y asimetría facial con ptosis palpebral izquierda. Por lo frecuente de las craneosinostosis, es necesario ante la sospecha efectuar un diagnóstico precoz, con intervención oportuna para un mejor tratamiento y seguimiento multidisciplinario, permitiendo esto realizar un adecuado asesoramiento genético a estas familias.
DeCS: CRANEOSINOSTOSIS, ACROCEFALOSINDACTILIA, SINOSTOSIS
The Saethre-Chotzen syndrome is among the hereditary craniosynostosis that is characterized by an early unilateral or bilateral synostosis of the coronal sutures, facial asymmetry, ptosis and sometimes strabismus. The case of a 10-year-old female patient is presented with craniostenosisprovoking skull asymmetry, plagiocephaly and facial asymmetry with left palpebral ptosis. Because of the frequency of craniosynostosis, the early diagnosis is necessary to perform a timely intervention for an accurate treatment and multidisciplinary follow-up, in order to complete an adequate genetic counseling to these families.
DeCS: CRANIOSYNOSTOSIS, ACROCEPHALOSYNDACTYLY, SYNOSTS
INTRODUCCIÓN
El síndrome Saethre-Chotzen(SCS), también llamado acrocefalosindactilia tipo III, fue descrito por primera vez por el neurólogo noruego Haakon Saethreen 1931 y el psiquiatra alemán Fritz Chotzenen 1932. Esta enfermedad constituye una craneosinostosis de origen genético que se transmite de padres a hijos según un patrón autosómico dominante. 1
Se caracteriza porque los individuos afectados presentan un espectro variable de manifestaciones como son; sinostosis de las suturas coronales (o con menos frecuencia sagital, metópicas o lambdoideas) que tiene como resultado una forma anómala del cráneo, asimetría facial, línea de la frente baja, ptosis, estrabismo, estenosis del conducto lacrimal. Otras manifestaciones frecuentes son braquidactilia, dedos de los pies anchos, sindactília cutánea parcial del dedo 2º y 3º de la mano y pies, así como falange distal duplicada del primer dedo del pie 1,2. La inteligencia es normal en la mayoría de los casos, aunque los individuos con grandes deleciones son más propensos a tener retrasos en el desarrollo, así se han registrado algunos casos con retraso en el desarrollo de leve a grave.3
Este tipo de craneosisnostosis es causado por mutaciones o deleciones puntuales que afectan (o eliminan completamente) al gen TWIST1 es el único gen asociado con el síndrome Saethre-Chotzen, localizado en el brazo corto del cromosoma 7p21.3-p21.2, que codifica un factor de transcripción hélice-bucle-hélice básico responsable de la determinación y la diferenciación de la citocinesis 4. Las mutaciones de pérdida de actividad en este gen provocan la inducción de fusiones prematuras de las suturas craneales. Las mutaciones se detectan en cerca del 80% de los individuos afectados mediante una combinación de análisis de deleción/duplicación y análisis de secuencia. El análisis de la secuencia del exón 1 del gen TWIST1 (único que se traduce) detecta todas las mutaciones intragénicas en este gen. Estas deleciones génicas pueden causar fenotipos más graves, normalmente asociados a retrasos neurocognitivos significativos. Se han comunicado mutaciones en FGFR3, FGFR2 y TCF12 que producen síndromes de craneosinostosis que se solapan fenotípicamente con el SCS.5
]]> Se estima su prevalencia uno de cada 25.000 a 50.000 personas, aunque existen muchos pacientes no diagnosticados, debido a que con frecuencia los síntomas son muy leves y pasan inadvertidos.Por ser la craneosinostosis frecuente en la práctica médica es necesario realizar un seguimiento a los casos con sospecha clínica para realizar un diagnóstico precoz, para poder efectuar intervención oportuna. Considerándose muy importante llegar al diagnóstico etiológico de esta afección para poder brindar un adecuado asesoramiento genético a estas familias.
PRESENTACION DE CASO
Paciente femenina de 10 años de edad, remitida a consulta de genética clínica por asimetría facial y ptosis palpebral.
Antecedentes familiares: no refieren que ninguna alteración de cráneo, ni asimetría facial.
Antecedentes prenatales: no se refieren ninguno de interés.
Antecedentes perinatales: parto eutócico a las 39,5 semanas, peso 3210 gramos, talla 49, 5 cm, circunferencia cefálica (CC): 34 cm, apgar 8-9.
Antecedentes postnatales: buena ganancia de peso, con buen desarrollo pondoestatural y psicomotor, con sostén cefálico a los 4 meses, se sostuvo sentada a los 6 meses, gateo a los 8 meses, camino a los 11 meses y decía palabras cortas a los 12 meses, buen desarrollo en la etapa transicional.
Se valoro en consulta por el maxilofacial por la simetría facial y oftalmología por la ptosis palpebral y el estrabismo.
]]> Al examen físico se observa asimetría de cráneo (plagiocefalia) con tendencia a la braquicefalia, así como asimetría facial con frente amplia, hendiduras palpebrales asimétricas con ptosis palpebral izquierda, estrabismo convergente, desviación del puente nasal hacia ese mismo lado como se puede apreciar en la figura 1
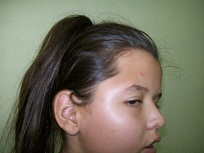
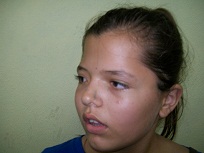
Figura 1. Características del cráneo y la facie de la paciente.
En el interrogatorio respondió acorde a su edad, mostrando capacidad intelectual normal.
Al examinar las extremidades se aprecia sindactília membranosa ligera entre el 2do y 3er dedo de ambos pies, así como clinodactília del 5to dedos de ambas manos, como se observa en la figura 2. ]]>
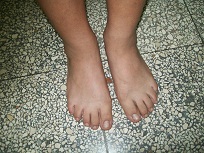
Figura 2. Particularidades de los pies de la paciente.
Se solicitó a la familia consentimiento informado, para examinarla y efectuarle radiografías de cráneo y tomarle fotos con el fin de ser publicarlas en revistas científicas, los cuales aceptaron firmando el mismo. Se confeccionó historia clínica genética, se revisó bibliografía actualizada, se estudiaron las radiografías donde no se observaron signos agudos hipertensión endocraneal, pero se confirma la plagiocefalia, discutiéndose el caso en colectivo, se llegó al diagnóstico clínico del síndrome de Saethre-Chotzen.
DISCUSION
Este síndrome pertenece al grupo de enfermedades llamadas acrocefalosindactilias, las principales manifestaciones consisten en anomalías en la forma del cráneoproducidas por un cierre prematuro de las suturas craneales (craneosinostosis). Esta alteración se produce durante la formación del cráneo, ocasionando plagiocefalia, braquicefalia, asimetría facial, que también se acompaña de malformaciones en las extremidades como clinodactíliay sindactília. 1,2
Esta craneosinostosis con patrón autosómico dominante, tiene un riesgo de recurrencia alto para la descendencia de estos enfermos, pero cuando no se recogen afectados en una familia y se presenta un caso aislado por lo general ocurre por una mutación novo, es decir una mutación nueva. Esta se trasmite con gran variabilidad de expresión, es subdiagnosticado en gran medida, pudiendo esta ser la causa de los pocos casos documentados en el país. 3
Existen laboratorios donde se cuenta con tecnología para realizar análisis de genética molecular para identificar mutación o deleción en gen TWIST1 que confirme el diagnóstico molecular de esta enfermedad. Aunque se ha publicado una familia con las típicas características fenotípicas del síndrome Saethre-Chotzen, sin mutaciones en el gen TWIST1 y sin embargo tenían una mutación p.Gln289Pro en el gen FGFR2, mutación frecuentemente reportada en otras craneosinostosis. Otro reporte de caso documento un rearreglo cromosómico nuevo entre los cromosomas 1, 4, 7 con microdelecion de 7p21.3-7p15.3, incluyendo el gen TWIST1, esto obliga a especular sobre la heterogeneidad genética que presenta esta enfermedad, que dificultaría el estudio de todas las mutaciones. 4,5
El diagnóstico pudiera efectuarse con formas sindrómicas de craneosinostosis, que cuando existen afectados en una familia se pudiera realizarse diagnóstico prenatal ultrasonográfico buscando craneosinostosis prematura. El diagnóstico diferencial es obligado con los síndromes que exhiben acrocefalosindactilia como el síndrome Apert que es una craneosinostosis que afecta sobre todo a sutura coronaria, lo que da lugar a una acrobraquicefalia muy acentuada con sindactília de manos y pies, membranosa y ósea entre 2do,3ro y 4to 6. También debe realizarse con el síndrome Crouzon, esta con alteraciones craneofaciales que definen clínicamente el cuadro con braquicefalia, turricefalia, abombamiento frontal, hipertelorismo y exoftalmos. El síndrome de Pfeiffer, que se define por la braquicefalia y los pulgares anchos, además debe diferenciarse de la sinostosis coronal unilateral aislada.7
]]> La capacidad intelectual es normal por lo general, aunque en algunas ocasiones existe ligero retraso mental. Aunque se han reportado microdelecion de en el brazo largo del cromosoma 4 (4q13.2-q13.3) y (7p15.3-p21.1) asociado con fenotipo síndrome Saethre-Chotzen, con discapacidad intelectual severay autismo. Esta paciente no tiene discapacidad intelectual, a diferencia de lo descrito por otros autores.8,9El diagnóstico se basa principalmente en la presencia de datos clínicos característicos. La tomografía de cráneo y las radiografías son útiles para caracterizar las anomalías del cráneo, clasificar la craneosinostosis y realizar diagnóstico de hipertensión intracraneal. En las radiografías de cráneo de esta paciente, se aprecia sinostósis de la sutura sagital pero no hay evidencias clínicas, ni radiográficas de hipertensión endocraneal.10
Este trastorno genético con un alto grado de variabilidad en la expresión fenotípica así como en la penetrancia, resultando difícil su identificación cuando no se realiza un adecuado seguimiento multidisciplinario, para poder evaluar por etapas cada signo clínico, para poder efectuar un diagnóstico precoz y una intervención oportuna, en función de un tratamiento y seguimiento adecuado, esto accederá a ejecutar un asesoramiento genético apropiado para las familias con afectados.
REFERENCIAS BIBLIOGRAFICAS
Wang JC, Nagy L, Demke JC. Syndromic Craniosynostosis. Facial Plast Surg Clin North Am. [Internet] 2016 Nov [Citado 3 de Ene 2017]; 24(4): [Aprox. 12p.]. Disponible en: http://www.sciencedirect.com/science/article/pii/S1064740616300633
Ko JM. Genetic Syndromes Associated with Craniosynostosis. J Korean Neurosurg Soc. [Internet] 2016 May [Citado 3 de Ene 2017]; 59(3): [Aprox. 4p.]. Disponible en: https://www.jkns.or.kr/journal/view.php?doi=10.3340/jkns.2016.59.3.187
Altiner Ş, Karabulut HG, Yararbaş K, Tükün A, Collet C, Kocaay P, et al. A novel TWIST1 gene mutation in a patient with Saethre-Chotzen syndrome. Clin Dysmorphol. [Internet] 2016 Nov 18. [Citado 3 de Ene 2017]. Disponible en: http://journals.lww.com/clindysmorphol/Citation/publishahead/A_novel_TWIST1_gene_mutation_in_a_patient_with.99627.aspx
Tahiri Y, Bastidas N, McDonald-McGinn DM, Birgfeld C, Zackai EH, Taylor J, et al. New Pattern of Sutural Synostosis Associated With TWIST Gene Mutation and Saethre-Chotzen Syndrome: Peace Sign Synostosis. J Craniofac Surg. [Internet] 2015 Jul [Citado 3 de Ene 2017]; 26(5): [Aprox. 3p.]. Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/26114524
Thakur AR, Naikmasur VG. A case of Robinow-Sorauf syndrome (Craniosynostosis-Bifid Hallux Syndrome): The allelic variant of the Saethre-Chotzen syndrome. Indian J Dent. [Internet] 2014 Apr [Citado 3 de Ene 2017]; 5(2): [Aprox. 3p.]. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4184321/
Massalska D, Bijok J, Kucińska-Chahwan A, Jamsheer A, Bogdanowicz J, Jakiel G, et al. Prenatal diagnosis of craniosynostosis (compound Saethre-Chotzen syndrome phenotype) caused by a de novo complex chromosomal rearrangement (1; 4; 7) with a microdeletion of 7p21.3-7p15.3, including TWIST1 gene--a case report. Ginekol Pol. [Internet] 2014 Jul [Citado 3 de Ene 2017]; 85(7): [Aprox. 5p.]. Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/25118508
Shimada S, Okamoto N, Nomura S, Fukui M, Shimakawa S, Sangu N, et al. Microdeletions of 5.5 Mb (4q13.2-q13.3) and 4.1 Mb (7p15.3-p21.1) associated with a saethre-chotzen-like phenotype, severe intellectual disability, and autism. Am J Med Genet A. [Internet] 2013 Aug [Citado 3 de Ene 2017]; 161A(8): [Aprox. 5p.]. Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.36027/full
Ketwaroo PD, Robson CD, Estroff JA. Prenatal Imaging of Craniosynostosis Syndromes. Semin Ultrasound CT MR. [Internet] 2015 Dec [Citado 3 de Ene 2017]; 36(6): [Aprox. 11p.]. Disponible en: http://www.sciencedirect.com/science/article/pii/S0887217115000542
![]() Elayne Esther Santana Hernández: Médica. Especialista de Segundo Grado en Medicina General Integral y Genética Clínica. Máster en Atención Integral al Niño Profesor Auxiliar. Investigador Agregado. Centro Provincial de Genética Médica de Holguín. Cuba. Si usted desea contactar con el autor de la investigación hágalo aqui
Elayne Esther Santana Hernández: Médica. Especialista de Segundo Grado en Medicina General Integral y Genética Clínica. Máster en Atención Integral al Niño Profesor Auxiliar. Investigador Agregado. Centro Provincial de Genética Médica de Holguín. Cuba. Si usted desea contactar con el autor de la investigación hágalo aqui