A propósito de un caso de gangliosidosis GM-2 tipo II: enfermedad de Sandhoff
A propos of a case of gangliosidosis GM. Type II. Sandhoff disease
Dra. Irelis González López,I Est. Enrique Marcos Sierra Benítez,IIEst. Mairiannys Quianella León Pérez,II Dra. Mayra Luz Leiva González,IDra. Samia Hernández Dreke,I Dr. Yadiel González FernándezIII
I Hospital Territorial Docente Dr. Mario Muñoz Monroy. Matanzas, Cuba.
II Filial Universitaria Dr. Eusebio Hernández Pérez. Matanzas, Cuba. ]]>
III Hospital Provincial Clínico Quirúrgico Docente José Ramón López Tabrane. Matanzas, Cuba.
RESUMEN
Las gangliosidosis son un conjunto de enfermedades hereditarias de almacenamiento lisosómico, debidas a un acúmulo de gangliósidos, sobre todo en las neuronas. La causa es la disfunción de alguna de las enzimas lisosómicas de la ruta de degradación de los gangliósidos. Existen varias formas de gangliosidosis, como son la GM1 y GM2. Se presentó el caso de una paciente de 33 años de edad, que había sido diagnosticada anteriormente de esclerosis lateral amiotrófica. Por varios síntomas presentados se le realizan una serie de exámenes complementarios, los cuales arrojan como resultado una gangliosidosis GM-2 tipo II o enfermedad de Sandhoff.
Palabras clave: gangliosidosis GM-2, enzimas lisosómicas, enfermedades de almacenamiento lisosómico.
ABSTRACT
Gangliosidosis are a group of hereditary diseases of lysosomal storage, due to an accumulation of gangliosides, especially in the neurons. The cause is the dysfunction of several lysosomal enzymes in the way of the gangliosides degradation. There are several forms of gangliosidesis, like GM1 and GM2. We present the case of a 33-years-old patient who was previously diagnosed with lateral amyotrophic sclerosis. Because of several symptoms he presented we carried out some complementary exams showing as a result a gangliosidosis GM-2 Type II or Sandhoff disease.
Key words: gangliosidesis GM-2, lysosomal enzymes, lysosomal storage disease.
INTRODUCCIÓN
Las gangliosidosis son un conjunto de enfermedades de almacenamiento lisosómico hereditarias debidas a un acúmulo de gangliósidos (un tipo de esfingolípidos), sobre todo en las neuronas. La causa es la disfunción de alguna de las enzimas lisosómicas de la ruta de degradación de los gangliósidos.
Existen varias formas de gangliosidosis:
Gangliosidosis GM1. Existe un acúmulo de gangliósido GM1 por déficit de ß-galactosidasa. Produce una enfermedad denominada gangliosidosis generalizada o pseudogargolismo, que se caracteriza por trastornos neurológicos, los niños no llegan al año de vida, visceromegalia, dolicomegalocefalia, macroglosia, retraso mental, sordera, enanismo, hepatomegalia, deformidades óseas, abdomen prominente, fascies característica.(1)
Gangliosidosis GM2. La forma más común es el acúmulo de gangliósidoGM2 por déficit de hexosaminidasa-A; provoca la enfermedad de Tay-Sachs, que afecta a las neuronas; el GM2 se acumula en ellas de forma precoz y severa, produciendo trastornos neurológicos. Cuando también se ve afectada la hexosaminidasa-B se acumula globósido, produciéndose una gangliosidosis GM2 generalizada o enfermedad de Sandhoff. La enfermedad de Tay-Sachs y la enfermedad de Sandhoff son males hereditarios que afectan el sistema nervioso central. Ambas enfermedades presentan los mismos síntomas pero son causadas por mutaciones (cambios) en diferentes genes.(2)
La enfermedad de Sandhoff o enfermedad de Sandhoff-Jatzkewitz es una enfermedad genética de almacenamiento lisosómico, autosómica recesiva heterógenea que afecta el sistema nervioso, cuya causa es la deficiencia en las enzimas β hexosaminidasa A y β hexosaminidasa B de los lisosomas teniendo como resultado la acumulación de esfingolípidos (gangliósidoGM2 y globósido) en el cerebro y otros órganos del cuerpo. La hexosaminidasa es necesaria para descomponer algunas sustancias grasas (gangliósidos GM 2) en las células del cerebro. Sin esta enzima, estas sustancias se acumulan y destruyen gradualmente las células del cerebro, hasta que todo el sistema nervioso central deja de funcionar.(3)
Muestra la sintomatología neurológica de la enfermedad de Tay-Sachs (retraso mental, ceguera, muerte prematura), pero con adicional compromiso visceral y una progresión mucho más rápida.
Estas enfermedades también presentan otras formas de aparición tardía, conocidas como enfermedad de aparición juvenil y adulta, según las características de los síntomas y la edad a la que aparecen. Mientras que los bebés con la enfermedad de Sandhoff clásica no producen la enzima hex A y B, las personas afectadas con las formas de aparición tardía producen cantidades muy pequeñas. Esto contribuye a explicar por qué los síntomas aparecen más tarde en una etapa posterior de su vida y son, en general, más leves que los de la forma clásica.(4)
]]> Los síntomas de la enfermedad de Sandhoff de aparición adulta (o enfermedad de Sandhoff crónica) son mucho más leves que los que caracterizan a las formas clásicas y juvenil. Los síntomas comienzan entre la adolescencia y los 35 años, pero pueden comenzar en la infancia. Por lo general, las personas afectadas no suelen perder la visión ni la audición. Algunas personas pueden perder ciertas habilidades mentales y tener problemas de memoria y comprensión. Los síntomas varían considerablemente en su severidad e incluyen, entre otros, tendencia a arrastrar las palabras al hablar, debilidad muscular, dolores musculares, temblores, andar inestable y, a veces, enfermedad mental. La expectativa de vida es variable y, en algunos casos, no se ve afectada.(2,4)La enfermedad de Sandhoff puede ocurrir en cualquier grupo étnico, pero es poco común. Los individuos que no son de origen judío tienen más probabilidades de ser portadores de las mutaciones genéticas que causan la enfermedad de Sandhoff que los de origen judío (una en 600 vs. una en 1 000).
La enfermedad de Sandhoff es causada por mutaciones en un gen del cromosoma 5 que codifica la enzima hex B. Ambas enfermedades son transmitidas por padres portadores de una de estas mutaciones. El portador no desarrolla la enfermedad. Sin embargo, cuando dos portadores deciden ser padres:(4)
-Existe una probabilidad del 25 % (una en cuatro) de que cualquiera de sus hijos herede una mutación del gen de cada uno de sus padres y desarrolle la enfermedad.
-Existe una probabilidad del 25 % (una en cuatro) de que el niño herede el gen normal de cada uno de sus padres. El niño no desarrollará la enfermedad ni será portador.
-Existe una probabilidad del 50 % (dos en cuatro) de que el niño herede un gen normal y uno anormal. El niño no desarrollará la enfermedad pero será portador al igual que los padres.
Si solo uno de los padres es portador del gen, ninguno de sus hijos tendrá la enfermedad, pero todos tendrán un 50 % de probabilidades de heredar la mutación del gen y ser portadores.
Normalmente se realiza una prueba de detección a los portadores antes o durante el embarazo en el caso de adultos pertenecientes a grupos en riesgo.(5)
Mediante las pruebas prenatales llamadas amniocentesis y muestra del villus coriónico (CVS) es posible diagnosticar esta enfermedad antes del nacimiento. En la amniocentesis, que generalmente se practica entre las semanas 15 y 20 del embarazo, el médico inserta una aguja en el abdomen de la madre para tomar una muestra del líquido que rodea al feto. Este líquido contiene células fetales que son analizadas para detectar la presencia de hex A o de hex B.
Si las pruebas prenatales detectan que solo falta la enzima hex A, el bebé tendrá la enfermedad de Tay-Sachs clásica. Si faltan las dos enzimas (hex A y hex B), el bebé tendrá la enfermedad de Sandhoff clásica.(6)
]]> Motiva la presentación del caso su rara frecuencia de aparición, pues constituye uno de los escasos casos diagnosticados en nuestro país y uno de los pocos en América Latina y del Caribe.
PRESENTACIÓN DEL CASO
Motivo de Ingreso: falta de aire.
Historia de la enfermedad actual: paciente YMP, con historia clínica no. 653860, femenina, blanca, de 33 años de edad, con antecedente patológico personal de esclerosis lateral amiotrófica. Por tal motivo es atendida en el Instituto de Neurología, en La Habana, con tratamiento farmacológico de ácido fólico, polivit y clordiazepóxido. Cuatro días antes del ingreso comienza con fiebre de 38 °C, precedida de escalofríos, dolor en punta de costado derecho y tos húmeda con expectoración mucopurulenta. Seguidamente comienza con dificultad respiratoria constante, que empeoraba progresivamente y no guardaba relación con el decúbito. Por estas razones acude a consulta y se decide su ingreso en la sala de Medicina Interna del Hospital Territorial Docente Dr. Mario Muñoz Monroy, del municipio de Colón, provincia de Matanzas, el 22 de octubre de 2013, con el presuntivo diagnóstico de una neumonía de base pulmonar derecha.
Examen físico: En el sistema respiratorio se constató expansibilidad torácica disminuida a la inspección y se comprueba con la maniobra de vértice-base, disnea, tiraje supraclavicular, supraesternal e intercostal, sonoridad pulmonar disminuida, murmullo vesicular disminuido globalmente, crepitantes en base pulmonar derecha, FR: 29. Ruidos cardiacos taquicárdicos (110 lat/min), T/A: 100/60 mm/Hg. En el Sistema Neurológico se observó afasia motora, hipotonía y pérdida de la motilidad de los cuatro miembros, temblores en reposo, arreflexia osteotendinosa y signo de Babinski. Se constató hepatomegalia y esplenomegalia a la palpación de abdomen.
Exámenes complementarios:
Gasometría arterial: acidemia respiratoria.
Ph: 7.337, PCO2: 56.0 mmHg, cHCO3 mmol/L: 30.7, PO2 mmHg: 50.6, E.B: 3.7 mmol/L.
Ionograma: hipopotasemia.
]]> Na: 141mmol/L, Cl: 99.6 mmol/L, K: 2.74 mmol/L, Ca: 1.129 mmol/L.Hemograma: Hemoglobina (Hb): 11.0 g/L; Hematocrito (Hto): 0.33U/L; Eritrosedimentación: 103 mm/h.
Leucograma: 13,4 x 109 L (leucocitosis)
Stab o células inmaduras: 0.02 x 109 L. Segmentados: 0.88 x 109 L.
Eosinófilos: 0.04 x 109 L. Monocitos: 0.06 x 109 L.
Glicemia: 3.3 mmol/L.
Creatinina: 43 mmol/L
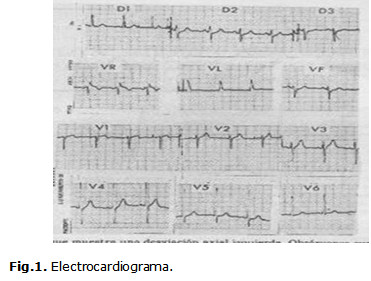
Electrocardiograma: mostró ritmo sinusal, regular (equidistancia entre todos los QRS), eje eléctrico intermedio (0°-90°), posición del corazón semihorizontal, frecuencia cardiaca de 110 latidos por minutos, microvoltaje en la derivación de AvL. (Fig. 1)
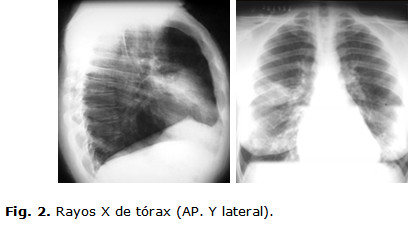
Rayos X de tórax: radiopacidad en forma triangular con base a la periferia, sombra densa, homogénea, bien delimitada en base pulmonar derecha. (Fig. 2)
]]>
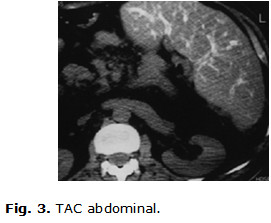
TAC abdominal: mostró hígado y bazo aumentado de tamaño (hepatomegalia y esplenomegalia), con pequeñas calcificaciones en el bazo. (Fig. 3)
Planteamiento sindrómico: síndrome de condensación inflamatoria lobar; síndrome neurológico tetrapléjico.
Planteamiento nosológico: neumonía de base pulmonar derecha; esclerosis unilateral amiotrófica.
Impresión diagnóstica: esclerosis unilateral amiotrófica agudizada por neumonía de base derecha.
Diagnóstico diferenciales: Enfermedad de Creutzfeldt-Jakob o síndrome de los priones, atrofia muscular espinal, esclerosis lateral primaria, Enfermedad de Kennedy o atrofia muscular progresiva espinobulbar.
La paciente durante su estadio hospitalario en la sala de Medicina Interna del Hospital Territorial Docente Dr. Mario Muñoz Monroy (3 días) evolucionó desfavorablemente a pesar de la suministración de antibióticos (cefuroxima) para la neumonía extrahospitalaria que presentó. Empeoró el cuadro respiratorio a pesar de la administración de oxígeno y continuaba con el déficit neurológico motor referido en el examen físico, por lo que se decide realizar rayos X de tórax evolutivo, el cual resultó negativo. La paciente presentó como único síntoma positivo la polipnea, la que se interpretó como resultado de una lesión del centro respiratorio producto a su enfermedad de base, se le indica complementarios de urgencia (gasometría, ionograma, leucograma, glicemia) y se le indica sonda levín, pues no deglute los alimentos. Debido a su dependencia de ventilación artificial para su respiración se decide su traslado para la Unidad de Cuidados Intensivos del mismo hospital. Allí se procedió a la ventilación mecánica de la paciente, debido a la gran debilidad de los músculos respiratorios y la depresión del centro respiratorio que presentaba.
Durante su estadio hospitalario se le realiza una extracción de sangre y se envía para el Instituto de Neurología y Neurocirugía, donde se detecta la deficiencia de la enzima hexosaminidasa A y B, por lo que es diagnosticada por el departamento de Neurogenética como una gangliosidosis GM-2 tipo 2 o enfermedad de Sandhoff de aparición tardía.
También puede utilizarse una muestra de sangre para realizar pruebas genéticas basadas en el ADN, y así detectar mutaciones conocidas en los genes hex A o hex B. Estas pruebas pueden ser útiles cuando son inciertos los resultados de la prueba común de portador explicada anteriormente.
]]> Después de 9 días de evolución desfavorable en la sala de Cuidados Intensivos, la paciente fallece producto de un paro cardiorrespiratorio, como complicación de su patología de base.
DISCUSIÓN
La fisiopatología neurológica mostrada por la paciente (tetraplejía) estuvo provocada por la acumulación excesiva de esfingolípidos y globósidos, que produjeron deterioro de las vainas de mielina que envolvían los axones de los haces piramidal y geniculado, provocando la imposibilidad de realizar cualquier movimiento, a su vez queda afectado el centro respiratorio (CR) como una estructura más del sistema nervioso. Este hecho no fue favorable, pues esta depresión del CR además de provocar un ambiente hipercápnico en el medio interno (acidosis), que favorece la proliferación de infecciones como la neumonía con que fue recibida, también evita la movilización de secreciones y expulsión de las mismas, lo que conlleva a un éxtasis que condensa aún más el proceso inflamatorio. La visceromegalia es muy común en todos los tipos de gangliosidosis, pues el marcado déficit enzimático de hexosaminidasa A y B provocan un acúmulo patológico en la matriz extracelular e incluso en el citoplasma de muchas células, aumentando así su tamaño microscópico, las más comúnmente afectada a parte de las nerviosas son las hepáticas, esplénicas y cardiacas.(6,7)
Actualmente, no hay tratamiento que impida que estas enfermedades sigan su curso. Solo se puede hacer que los niños afectados lleven la vida más cómoda posible, ofreciéndoles todo tipo de apoyo.(8)
Los investigadores están estudiando si los transplantes de células madre (a veces llamados transplantes de médula ósea) pueden ayudar a los pacientes afectados con la enfermedad de Tay-Sachs o de Sandhoff clásicas. Las células madre son células sanguíneas inmaduras que producen todos los demás tipos de células sanguíneas. Estas células se obtienen de la sangre del cordón umbilical o de la médula ósea de un donante. Lamentablemente, los transplantes de células madre aún no han logrado detener o revertir el daño cerebral causado por las enfermedades de Tay-Sachs o de Sandhoff y presentan un alto riesgo de muerte en los bebés afectados.(2,5,7)
Los médicos también están investigando la eficacia de tratamientos con medicamentos (incluido un medicamento llamado miglustat, aprobado por la Dirección de Alimentos y Medicamentos de los Estados Unidos para el tratamiento de un trastorno relacionado) para reducir la acumulación de sustancias grasas en las células cerebrales en personas afectadas con estas enfermedades.
Los investigadores subvencionados por March of Dimes han contribuido a la identificación de varias mutaciones del gen hex A causantes de las formas de aparición tardía de la enfermedad de Tay-Sachs. La información sobre mutaciones específicas permite realizar un mejor diagnóstico y estudios de portador para todas las formas de esta enfermedad.
La gangliosidosis GM2 y los errores congénitos del metabolismo que ocasionan detención y regresión del neurodesarrollo son un reto para la comunidad científica, porque requieren de un diagnóstico oportuno para informar y orientar adecuadamente a la familia.(6-11)
]]> REFERENCIAS BIBLIOGRÁFICAS
1- Suzuki Y, Sakuraba H, Oshima A. Galactosidase deficiency (Galactosidosis): GM 1 Gangliosidosis and Morquio B Disease. En: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York: McGraw-Hill; 2012 [citado 16 Dic 2014]. Disponible en: http://www.ommbid.com/OMMBID/the_online_metabolic_and_molecular_bases_of_inherited_disease /b/abstract/part16/ch151
2- Pozo Alonso AJ, Pozo Lauzán DR, Estol de la Guardia N, Menéndez Sainz C. Enfermedad de Tay-Sachs. Rev Cubana Pediatr [Internet]. 2014 [citado 17 Dic 2014];86(4):529-34. Disponible en: http://scieloprueba.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75312014000400014&nrm=iso
3- Menéndez Sainz MC. Diagnóstico enzimático de las enfermedades de almacenamiento lisosomal: experiencia de 20 años [tesis para optar por el grado de Doctor en Ciencias de la Salud]. La Habana: Universidad de Ciencias Médicas de La Habana; 2012 [citado 17 Dic 2014]. Disponible en: http://tesis.repo.sld.cu/423/1/MenendezSainzL.pdf
4- Uribe A. Hexosaminidasas y enfermedades neurodegenerativas. Iatreia [Internet]. 2011 [citado 18 Dic 2014];23(4). Disponible en: http://aprendeenlinea.udea.edu.co/revistas/index.php/iatreia/article/viewArticle/8195
5- Neufeld EF, Muenzer J. The Mucopolysaccharidoses. En: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York: McGraw-Hill; 2013 [citado 16 Dic 2014]. Disponible en: http://www.ommbid.com/OMMBID/the_online_metabolic_and_molecular_bases_of_inherited_disease/b /abstract/part16/ch136
6- Caciotti A, Garman S, Rivera Y, Procopio E, Catarzi S, Ferri L, et al. GM 2 gangliosidosis and Morquio B disease: an update on genetic alterations and clinical findings. Biochim Biophys [Internet]. 2011 [citado 17 Dic 2014];1812(7). Citado en PubMed; PMID: 21497194.
7- d'Azzo A, Andria G, Strisciuglio P, Galjaard H. Galactosialidosis. En: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York: McGraw-Hill; 2010 [citado 18 Dic 2014]. Disponible en: http://www.ommbid.com/OMMBID/the_online_metabolic_and_molecular_bases_of_inherited_disease/b /abstract/part16/ch152
8- Hernández García I, Seiglie Díaz F, Campos Hernández D, Marrón Portarles L, Díaz González JL, Carmona Padrón O. Gangliosidosis generalizada tipo 1. Rev Cubana Pediatr [Internet]. 2014 [citado 16 Nov 2014];86(1):103-7. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75312014000100013&nrm=iso
9- Jeyakumar M, Thomas R, Smith E, Smith D, van der Spoel A, Hugh Perry V, et al. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain [Internet]. 2003 [citado 18 Dic 2014];126(4):974-87. Disponible en: http://brain.oxfordjournals.org/content/126/4/974
10- Mahuran DJ. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease [Internet]. 1999 [citado 18 Dic 2014];1455(2-3):105-38. Disponible en: http://www.sciencedirect.com/science/article/pii/S0925443999000745
11- Clarkea JT, Mahurana DJ, Satheb S, Kolodnyb EH, Rigata BA, Raimana JA, et al. An open-label Phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay–Sachs or Sandhoff variants). Molecular Genetics and Metabolism [Internet]. 2011 [citado 18 Dic 2014];102(1):6-12. Disponible en: http://www.sciencedirect.com/science/article/pii/S1096719210003458
Recibido: 22 de diciembre de 2014.
Aceptado: 2 de abril de 2015.
Dra. Irelis González López. Hospital Territorial Docente Dr. Mario Muñoz Monroy. Martí final. Colón. Matanzas, Cuba. Correo electrónico: irelisgl.mtz@infomed.sld.cu
]]>