Descriptores DeCs: SÍNDROME DE FRAGILIDAD DEL CROMOSOMA X/genética; GENÉTICA BIOQUÍMICA; SOUTHERN BLOTTING/métodos; REACCIÓN EN CADENA POR POLIMERASA/métodos; CITOGENÉTICA.
Pocas enfermedades genéticas han despertado tanto interés como el síndrome Frágil X.
Desde que en 1943 Martin y Bell1 reportaron claras evidencias de herencia recesiva ligada al cromosoma X en una familia en la que los varones presentaban retraso mental, pasando por el reporte de Lubs en 19692 acerca de la relación entre el sitio frágil en el extremo distal de los brazos largos del cromosoma X, hasta la caracterización molecular del gen FMR-1,3 han transcurrido apenas 48 años.
Con los conocimientos actuales comienza una nueva etapa de investigaciones destinada a dilucidar el defecto básico que media entre el conocimiento molecular de esta segunda causa genética de retraso mental (RM).
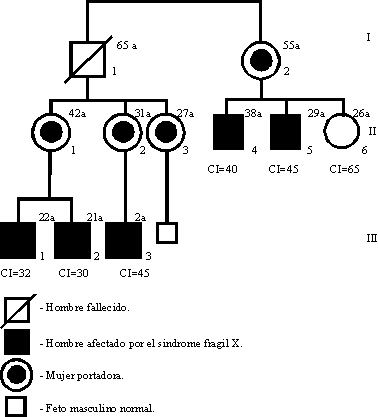
Nuestro propósito con este trabajo ha sido el de analizar la correlación existente entre las manifestaciones clínicas previamente delineadas, porcentaje de fragilidad Xq27.3 y la caracterización molecular del gen FMR-1, en 5 varones afectados y 4 mujeres portadoras obligadas de 3 generaciones de una familia registrada en el servicio de Genética Clínica del Hospital Pediátrico Docente "Juan Manuel Márquez".
Características clínicas físicas:
Volumen testicular (mL) = p /6 x l x a
donde: l = longitud del testículo y
a = ancho del testículo.
Se consideró macroorquidismo cuando el volumen testicular excedió los 25 mL en el adulto y los 2 mL en los prepuberales.6
La severidad de RM de los miembros de la familia se obtuvo mediante las pruebas psicométricas realizadas en el Centro de Orientación y Diagnóstico y se añadió una valoración cualitativa al tener en cuenta las esferas que afectan de manera peculiar a los pacientes según estudios neuropsicológicos reportados en la literatura médica7 y se valoró el desarrollo del lenguaje, la comunicación verbal y extraverbal, relaciones sociales, habilidades adquiridas y conducta general.
Las láminas se trataron con tripsina y coloreadas con giemsa para la obtención de bandas G (GTG). ]]>
Una vez excluidas otras aberraciones cromosómicas se procedió al análsis de 100 metafases para la detección del sitio frágil Xq27.3. Las muestras se enviaron al departamento de genética clínica de la Universidad de Erasmus en Rotterdan, Holanda, donde se extrajo el ácido desoxinucleótico (ADN) por métodos convencionales y se procesaron las muestras con 2 métodos directos para la caracterización del gen FMR-1:
| | |||||||
| | ]]> Neuropsicológicas | ||||||
| | | | | | | | |
| | ]]> Facie no específica | | | | | | |
| | | | ]]> 4/5 | | | | |
| | | | | | ]]> CI inferior | | |
| | | | | | | | ]]> 4/4 |
| | | | | | | | |
| | ]]> Macroorquidismo | | | | | | |
| | | | | ]]> 7 | | | |
| | | | | | |||
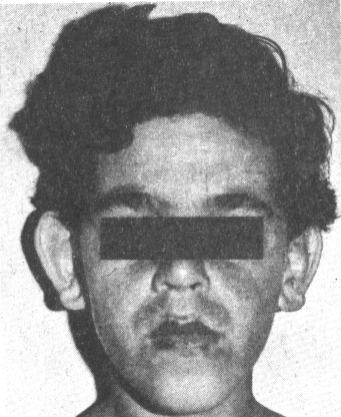 FIGURA 2. Características faciales en uno de los varones estudiados, donde se aprecia cara alargada, orejas grandes y mentón prominente.
FIGURA 2. Características faciales en uno de los varones estudiados, donde se aprecia cara alargada, orejas grandes y mentón prominente.
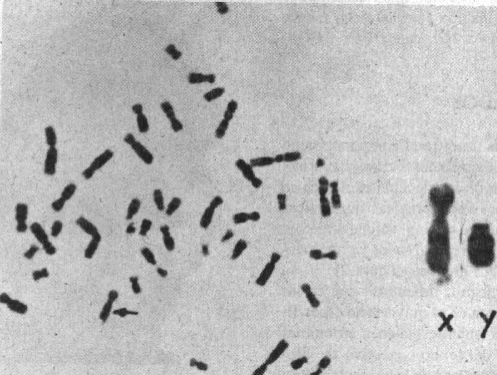
En la figura 3 se muestran las características del sitio frágil Xq27.3. Los estudios moleculares y citogenético de la portadora II-2 y de su hijo afectado III-3 (figura 1) se realizaron simultáneamente. El hombre I-1 fue estudiado citogenéticamente antes de su fallecimiento. Exceptuando a la mujer II-3 con 1 % de sitio frágil, el resto de las mujeres analizadas así como el hombre I-1 no mostraron sitio frágil Xq27.3. Los varones II-4 y II-5 tuvieron 3 y 2 % respectivamente y los valores más altos fueron vistos en III-1, III-2 y III-3 con frecuencias de 19,17 y 15 % de sitio FRAXA.
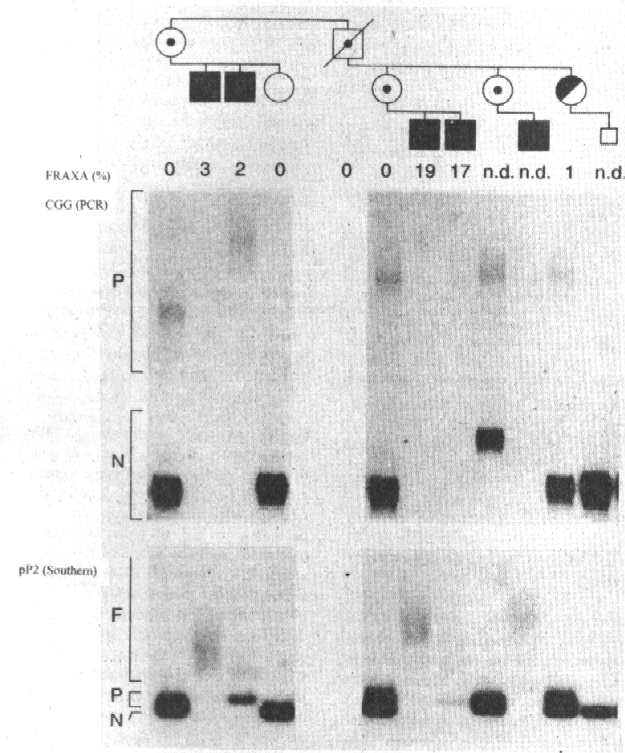
La caracterización molecular aparece en la figura 4, en la que se destaca en el diagnóstico prenatal de transmisión del cromosoma X normal de la portadora II-3 a su feto.
La correlación clínica citogenética y molecular se expresa en la figura 5 en la que se evidencia un incremento de severidad de las manifestaciones clínicas, así como de porcentaje de fragilidad en correspondencia con el paso de la mutación en las generaciones estudiadas.

Los defectos propios del tejido conectivo como paladar alto y pectus excavatum o carinatum se observaron solamente en los varones afectados de la tercera generación, hecho éste no correlacionado con la edad y sí probablemente con la magnitud de la mutación completa. La hiperlaxitud articular es mucho más generalizada para todos los valores afectados y también en dependencia de la edad.5 ]]>
Las manifestaciones neuropsicológicas estudiadas son los elementos más significativos del síndrome. El RM aunque con CI inferior a 50, es más severo en los varones de la tercera generación, y se correlaciona con el porcentaje de fragilidad incrementada en estos pacientes, que por otra parte, como se ha reportado9 se hace evidente cuando el número de repeticiones CGG es superior a 200, debido posiblemente a la baja disponibilidad de citidina en medios deficientes de folato que interfieren la síntesis in vitro de las expansiones CGG.10 De ser así, predictivamente la familia estudiada, la expansión de la mutación completa sería mucho mayor en los varones de la tercera generación que en los de la segunda.Aunque la fisiopatología del síndrome no se ha confirmado, se sabe que cuando la elongación de CGG de la mutación es muy grande las regiones CpG adyacentes se hipermitilan y afectan a la transcripción del gen y a su vez la síntesis de la proteína que ésta codifica,11 lo que pudiera ser la ausencia de esta proteína la responsable de las manifestaciones neuropsicológicas descritas.11
A su vez el incremento de repeticiones CGG depende del tamaño de la premutación y el sexo del portador. Las premutaciones que transmite un hombre portador sano no varían al ser transmitidas a sus hijos, pero éstas se pueden incrementar notablemente al ser transmitidas a sus nietos.12
En esta familia esto sería otra explicación de la severidad clínica en los pacientes de la tercera generación respecto a la segunda, pues la mujer I-2 presenta una premutación de 80 repeticiones mientras que en las portadoras obligadas de la generación III la premutación es de 90 repeticiones para incrementarse durante la ovogénesis el riesgo de pasar a una mutación completa mucho mayor que la de los varones de la generación II.13
La epilepsia ha sido reportada y estudiada por otros autores al describir las anormalidades neurológicas del síndrome (Rodríguez H, Lantigua A, Guerra D. Síndrome frágil X en 35 varones con retraso mental inespecífico [trabajo de terminación de residencia de Genética Clínica] Centro Nacional de Genética Médica, ISCM-H, 1987). Se ha reportado correlación entre las convulsiones y el porcentaje de sitio frágil Xq27.3.11 En esta familia sólo un varón de la III generación no ha presentado epilepsia, lo que sugiere que este síntoma tal vez no dependa solamente de la mutación.
A pesar de que el hombre I-1 no pudo ser caracterizado molecularmente, el hecho de tener una hermana y todas sus hijas portadoras evidencian que él es un hombre transmisor sano. El 20 % de los varones portadores son fenotípicamente también normales, sus hijas son portadores y fenotípicamente también normales, mientras que sus nietos tendrán alto riesgo (40 %) de presentar el síndrome.14
Aunque no es el objetivo de este trabajo es importante señalar que la caracterización molecular del gen es de extraordinario valor para el diagnóstico prenatal, sobre todo si, como en este caso el feto varón hereda el cromosoma X normal de la madre.
Podemos concluir con que las manifestaciones físicas y neuropsicológicas en los varones afectados son más constantes y se pueden relacionar con la expresión de diferentes elongaciones CGG en la mutación completa del gen FMR-1.
En esta familia se puede correlacionar entre expresión de FRAXA y la elongación de la mutación completa FMR-1 al apoyar la idea de correspondencia entre ésta y la baja disponibilidad de citidina por la deficiencia de folato del medio de cultivo utilizado, que interfiere en el completamiento de la síntesis, in vitro de la expansión CGG y que se evidencia microscópicamente como un gap o ruptura al nivel de Xq27.3. cuando existen más de 200 repeticiones.
El fenotipo de las portadoras en nuestro estudio no permite establecer una correlación específica. El perfil neuropsicológico es susceptible de sobrelapamiento con otras posibles alteraciones genómicas o por factores ambientales psicofamiliares. La hija (II-6) de la mujer I-2 comparten fenotipos similares y genotipos diferentes, mientras que I-2 es heterocigótica, II-6 presenta 2 cromosomas X con el rango normal de repeticiones CGG.
Dra. Aracely Lantigua Cruz. Departamento de Genética Clínica. Centro Nacional de Genética Médica, ISCM-H, Calle 146 y Avenida 31, Cubanacán, municipio Playa, Ciudad de La Habana, Cuba.
1 Doctora en Ciencias Médicas. Especialista de II Grado en Genética Clínica. Profesora Titular. Jefa del Departamento de Genética Clínica, Centro Nacional de Genética Médica.
2 Especialista de I Grado en Genética Clínica.
3 Investigadora del Departamento de Genética Clínica, Universidad de Erasmus. ]]>