Se describe en una niña de 6 meses de edad, una enfermedad metabólica del tipo mucolipidosis II. Al examen físico se constató facies tosca, marcada hiperplasia gingival, cuello corto, deformidad torácica, piel gruesa y firme y retardo marcado del desarrollo psicomotor. El inicio temprano de la enfermedad, la excresión de oligosacáridos en la orina y el típico comportamiento de las enzimas lisosomales elevadas en sueron permitieron llegar a este diagnóstico.
Descriptores DeCS: MUCOLIPIDOSIS.
Las mucolipidosis (MLP) son un grupo de errores congénitos del metabolismo con acúmulo de mucopolisacáridos ácidos, esfingolípidos y glicolípidos en células mesenquimatosas y viscerales.1 Se clasifican en 4 grupos de acuerdo con el déficit enzimático que las determinan y el desarrollo clínico de la enfermedad.1,2 Suelen confundirse con las mucopolisacaridosis, de las cuales se diferencian por la ausencia de mucopolisacariduria. La MLP-II o enfermedad de células I fue descrita por Leroy y Demars en 1967.3 Su comportamiento bioquímico -deficiencia de enzimas lisosomales en cultivos de fibroblastos con actividad de las mismas enzimas elevadas en suero- es similar al de la MLP-III,1-5 pero se distingue clínicamente de ésta por el comienzo más temprano. Este reporte constituye la primera comunicación de esta entidad en nuestro medio.
MÉTODOS
PRESENTACIÓN DEL CASO
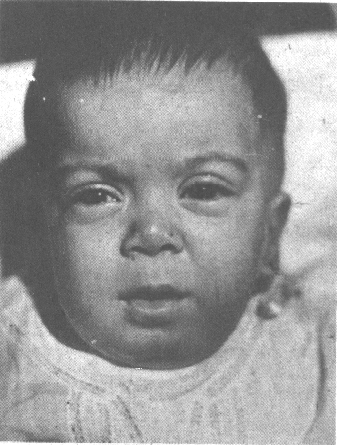
Paciente femenina, blanca, de 6 meses de edad, nacida de embarazo normal y parto fisiológico a las 37,5 semanas. Su peso al nacer fue de 2,3 kg. La madre tenía 24 y el padre 27 años al nacimiento de la niña; no eran consanguíneos y ambos procedían de 2 provincias localizadas en la parte oriental de Cuba. Se ingresa para estudio por retardo del desarrollo psicomotor y posible escleredema. Se remite a nuestra especialidad y al examen físico constatamos los siguientes signos: peso: 6 kg; talla: 64,5 cm; valoración nutricional: 10-25 percentil. Macrocráneo ligero. Turri-braquicefalia. Facies tosca con frente amplia, párpados hinchados, ojos saltones, epicantus, hipertelorismo, puente nasal deprimido, mejillas gruesas, narinas antevertidas, filtrum poco profundo, marcada hiperplasia gingival y de los rebordes alveolares; boca entreabierta; cuello muy corto (fig. 1). Pectus carinatus, cifosis dorso-lumbar; abdomen plano, depresible, no visceromegalias. Limitación de la movilidad articular en hombros, codos, rodillas y dedos de las manos. Muñecas engrosadas. Piel gruesa e indurada. Ruidos cardíacos rítmicos y bien golpeados. No sostiene la cabeza en decúbito prono. No se voltea. No se sostiene sentada sola. No gorjea. Hipotonía de tronco y extremidades. Cuadriparesia. Hiporreflexia osteotendinosa generalizada. Fondo de ojos normal.
FIGURA 1. Facies de la paciente a los 6 meses de edad.
RESULTADOS
Resultados de investigaciones realizadas:
a) Estudios radiológicos:
Macrocráneo.
Tórax estrecho. Costillas anchas.
Cuerpos vertebrales altos.
Metáfisis anchas e irregulares del extremo distal de cúbitos radios, fémures y proximal de las tibias.
]]>
Coxa valga.
Metacarpianos cortos, anchos e irregulares; punteagudos en sus extremos proximales.
b) Estudios ultrasonográficos:
Abdomen: No alteraciones viscerales.
Ecocardiograma: Normal.
c) TAC de cráneo simple:
Atrofia cortical y dilatación del sistema ventricular.
d) Electroencefalograma:
Trazado asimétrico con disminución de voltaje en hemisferio izquierdo. Ritmo de base Theta 4 Hz y delta 3 Hz.
Tamisaje metabólico en orina: a) Prueba de azul de toluidina: normal. ]]>
b) Prueba de azul alcián: 2 411 mg MPS/g de creatinina. c) TLC de aminoacidos: aminoaciduria. d) TLC de carbohidratos: azúcares reduc-tores y oligosacáridos. e) TLC de oligosacáridos: oligosacariduria. f) Prueba de Benedict: negativa. g) Prueba de Selivanoff: negativa.
DISCUSIÓN
La sospecha diagnóstica de una enfermedad metabólica la tuvimos desde el inicio, por la facies tosca y el retardo del desarrollo psicomotor. La facies presentaba un estrecho parecido con la de las mucopolisacaridosis, pero el comienzo de las manifestaciones clínicas fue evidentemente mucho más temprano que en aquéllas. Otro signo clínico importante fue la severa hiperplasia gingival, por la que la paciente no podía cerrar la boca. Sus encías hacían contacto en la región posterior, y provocaban la apertura de la región anterior de forma permanente. Es al parecer un signo importante de la enfermedad. Galili y colaboradores la describen como una hiperplasia extensa de las encías, que se registra poco después del nacimiento; interfiere con la masticación y precede y obstaculiza la erupción de la mayoría de los dientes.6 La oligosacariduria intensa fue la primera evidencia bioquímica de que se trataba de una oligosacaridosis. También se sospecharon otras causas metabólicas de facies tosca por los estudios cromatográficos en orina y los enzimáticos en leucocitos.1,2,7
Las enzimas lisosomales elevadas en suero permitieron confirmar el diagnóstico de MLP-II en la paciente. Las más elevadas fueron la betaglucosidasa, la arilsulfata A, las arilsulfatasas A,B,C, la betagalactosidasa, la alfa-L-iduronidasa y la beta-glucuronidasa (fig. 2). Las arilsulfatasas suelen considerarse buenos marcadores para el diagnóstico bioquímico de las MLP II y III.1,2,4,5 En ambos tipos de mucolipidosis el defecto enzimático es el mismo déficit de la enzima N-acetilglucosamina-fosfotransferasa. Esta fosfotransferasa cede fosfato a los residuos de manosa de la cadena de oligosacáridos de los precursores de las enzimas lisosomales. La manosa-6-fosfato constituye el grupo de reconocimiento para el receptor que facilitará la entrada de la enzima a la célula. Si hay déficit de fosfotransferasa las enzimas no serán "marcadas" y no podrán entrar al lisosoma; por lo que serán segregadas al medio extracelular. Este efecto secundario resultante en la elevación de las enzimas lisosomales en el suero ( o plasma), único de las MLP-II y MLP-III, se utiliza para el diagnóstico bioquímico con más frecuencia que las células I en fibroblastos cultivados o las determinaciones enzimáticas de la misma fofotransferasa en leucocitos y en fibroblastos, por razones obvias de accesibilidad e idoneidad del material biológico (suero) y de la prueba. Las variaciones clínicas entre ambas mucolipidosis se deben a la homocigocidad de diferentes genes mutantes al locus 4q21-q23.3,5,8,9
FIGURA 2. Enzimas lisosomales en suero
La MLP-II ha sido descrita fundamentalmente en poblaciones caucásicas y asiáticas.10-12 La prevalencia al nacer en una población franco-canadiense fue de 1/6 184 nacidos vivos y la de portadores de 1/39.10Lemaitre y colaboradores13 distinguen 2 etapas de la enfermedad: la etapa A, temprana o neonatal, desde el nacimiento hasta los 2 meses de edad y que se caracteriza por la desmineralización y trabeculación sin diferenciación entre las zonas cortical y medular del hueso, sin disturbio del metabolismo fosfocálcico, por lo que los diagnósticos diferenciales se deben realizar con el hiperparatiroidismo, el raquitismo y la osteomielitis. En casos con deformidades congénitas se ha confundido con osteogénesis imperfecta14 y en los que han presentado punteado de las epífisis con aquellos síndromes en los que éstas suelen aparecer.5 En la etapa B o tardía, que es en la que se diagnostican la mayoría de los casos, ocurre varios meses después y los signos esqueléticos son de disostosis múltiple severa. No se conoce mucho acerca de la afectación del sistema nervioso central; sin embargo, la cardiovascular está bien caracterizada.15,16 A nosotros la paciente nos impresionó mucho más retardada desde el punto de vista motor que en su capacidad mental. Esto ha sido señalado por otros autores.10-12,15Beck y colaboradores reportaron la variabilidad inter e intrafamiliar de la MLP-II. Como signos clínicos permanentes encontraron el nanismo, la facies tosca y el retardo mental; la variabilidad se hizo más evidente con respecto a la edad de comienzo, la afectación visceral y los hallazgos radiológicos. Esta variabilidad clínica en pacientes con genotipos idénticos no es rara en las enfermedades lisosomales y se atribuye al efecto de proteínas activadoras específicas que pueden modular la actividad enzimática, al pH dentro de los lisosomas y otros factores ambientales todavía desconocidos.15 ]]>
La enfermedad es fatal, rápidamente progresiva en algunos y más estable en otros, y la muerte ocurre entre los 2 a 5 años de vida, generalmente por fallo cardíaco durante alguna infección.1,11,12
Aunque los padres de la paciente no son consanguíneos sí consideramos de interés que ellos procedan de una zona de Cuba en la que hemos descrito pacientes con enfermedades lisosomales anteriormente desconocidas en nuestro medio.7 La importancia de llegar a este diagnóstico relativamente temprano, permitió advertir a una pareja de padres jóvenes acerca de los riesgos de recurrencia por tratarse de una afección autosómica recesiva,2 en la cual sólo es posible la detección de portadores por estudios moleculares.
SUMMARY
A metabolic disease knwon as Mucolipidosis II which affects a six-monts old girl is described in this work. Facies tosca, marked gingival hyperplasia, short neck, thorax deformity, thick and firm skin, and marked psychomotor retardation were detected when the girl was physically checked. The early onset of the disease, the oligosacharide excretion in urine and the typical behaviour of raised serum lysosomal enzimes levels made this diagnosis possible.
Subject headings: MUCOLIPIDOSIS.
REFERENCIAS BIBLIOGRÁFICAS
Gaaljard H. Genetic metabolic disease. 1ra ed. Amsterdam: Elsevier;1980:132-47.
Mc Kusick V. Mendelian Inheritance in Man. Catalogs of autosomal dominant, autosomal recessive and x linked phenotypes. Eleven Edition. Baltimore: Johns Hopkins University Press; 1994.
Leroy JG, Demars RI, Opitz JM. I cell disease. Birth Defects Orig Art Ser 1969;4;174-85.
Umehara F, Matsumoto W, Kuriyama M, Sukegawa K, Gasa S, Nitsuhiro O. Mucolipidosis III (pseudo-Hurler polydystrophy); clinical studies in aged patients in one family. J Neurol Sci 1997;146:167-72.
Mueller O, Wasmuth J, Murray J, Lozzio C, Lovrien E, Shows T. Chromosomal assignment of N-acetylglucosaminyl phospotransferase, the lysosomal hydrolase targeting enzyme deficient in mucolipidosis II and III. Cytogenetic Cell Genet 1983;46:664.
Gallili D, Yatzen S, Russell A. Massive gingival hyperplasia preceding dental eruption in I cell disease. Oral Surg 1974;37:533-9.
Menéndez I, Pozo D, Mar J, Castro J, Hernández M. Fucosidosis. Una enfermedad por almacenamiento lisosomal. Rev Cubana Pediatr 1993;65(2):138-44.
]]>
Ben-Yoseph, Potier M, Mitchell D, Park B, Melancon S, Nadler H. Altered molecular size of N-acetylglucosamine-1-phosphotransferase in I- cell disease and pseudo Hurler polydys-trophy. Biochem J 1987;248:697-701.
Vidgoff J, Rowe S, Stafford R, Buist N, Lovrien E. Localization of the gene for I cell disease (mucolipidosis II). Am J Hum Genet 1982;34:64.
De Braekeler M. Hereditary disorders in Saguenay-Lac-St Jean (Quebec, Canada). Hum Hered 1991;41:141-6.
Gilbert E, Dawson G, Zu Rheim, Opitz J, Spranger J. Pathological, histochemical, ultrastructural and biochemical observations in four cases. Z Kinderheilk 1973;114:259-92.
Yukata T, Ogawa M. I cell disease: clinical studies of 21 Japanese cases. Clin Genet 1985;28:207-15.
Lemaitre L, Remy J, Farriaux JP, Dhondt J, Walbaum R. Radiological signs of mucolipidosis II or I-cell disease. A study of nine cases. Pediatr Radiol 1978;7:97-105.
Michels V, Dulton R, Caskey C. Mucolipidosis II: unusual presentation with a congenital angulated fracture. Clin Genet 1982;21:225-7.
Beck M, Barone R, Hoffmann R, Kratzer W, Rakowsky T, Nigro F, et al. Inter-and intrafamilial variability in mucolipidosis II (I-cell disease). Clin Genet 1995;47:191-9.
Satoh Y, Sakamoto K, Fujibayashi Y, Uchigawa T, Kajiwara N, Hatano M. Cardiac involvement in mucolipidosis. Importance of Non-invasive studies for detection of cardiac abnormalities. Jpn Heart J 1983;24:149-59.
Recibido: 6 de junio de 1996. Aprobado: 28 de noviembre de 1997. ]]>
Dra. Ibis Menéndez Alejo. Hospital Pediátrico Docente "William Soler", San Francisco 10112, Altahabana, Ciudad de La Habana, Cuba.
1 Especialista de II Grado en Genética Clínica. Investigadora Agregada. Instructora de Genética. Departamento de Genética. Hospital Pediátrico Docente "William Soler". 2 Licenciada en Bioquímica. Investigadora Agregada. Laboratorio de Enzimas Lisosomales. Instituto de Neurología y Neurocirugía. 3 Especialista de I Grado en Neurología. Hospital Pediátrico Docente "William Soler". 4 Licenciada en Ciencias Biológicas. Especialista de Laboratorio. Centro Nacional de Genética Médica. ]]>19801ra132-471994Eleven19694174-851997146167-72198346664197437533-919936522138-441987248697-70119823464199141141-61973114259-92198528207-151978797-105198221225-7199547191-9198324149-59