Crecimiento intrauterino restringido como síntoma guía para el diagnóstico genético prenatal del síndrome de Wolf-Hirschhorn en gestación gemelar bicorial biamniótica
Restricted Intrauterine Growth as a Guiding Symptom for Prenatal Genetic Diagnosis of Wolf-Hirschhorn Syndrome in Bicorial Diamniotic Twin Gestation
Gonzalo Peláez Marín, Zoraida Frías Sánchez, Manuel Pantoja Garrido, Isabel Corrales Gutiérrez, José Luis Barroso Castro, José Antonio Gómez-Coronado Vinuesa
Unidad de Gestión Clínica de Obstetricia y Ginecología del Hospital Universitario Virgen Macarena de Sevilla. España.
El síndrome de Wolf Hirschhorn, también conocido como monosomía del brazo corto del cromosoma 4 (4p) o síndrome 4p-, es una rara enfermedad genética descrita por primera vez en el año 1961 por los doctores Cooper y Hirschhorn. El objetivo del trabajo es presentar un caso clínico sobre el síndrome de Wolf-Hirschhorn, que es un trastorno genético raro y aún bastante desconocido que cursa con múltiples anomalías morfológicas congénitas, así como con un retraso neurológico e intelectual de grado variable. La prevalencia de este síndrome es extremadamente baja, teniendo en cuenta que la cifra puede estar infraestimada, dada las pérdidas gestacionales precoces y la dificultad en el diagnóstico prenatal. Reportamos el caso de una paciente con gestación gemelar bicorial biamniótica tras un ciclo de FIV-ICSI, en el que al segundo gemelo se diagnosticó un Síndrome de Wolf-Hirschhorn, luego del estudio por una discordancia de pesos estimados y crecimiento intrauterino restringido de este segundo feto. El patrón clásico de presentación clínica se caracteriza por el desarrollo de alteraciones craneofaciales importantes, retraso en el crecimiento normal tanto prenatal como posnatal y deficiencia mental e intelectual de grado variable. El diagnóstico prenatal debe ser realizado por expertos. Puede sospecharse por un crecimiento intrauterino restringido, ya que se da en 80-90 % de los fetos con esta patología. Una vez diagnosticado, se recomienda el estudio genético de los padres, dado que hasta 15 % de los progenitores pueden padecer un reordenamiento cromosómico equilibrado en el brazo corto del cromosoma 4.
Palabras clave: síndrome Wolf-Hirschhorn; síndrome delección 4p; enfermedades congénitas; alteraciones cromosómicas.
ABSTRACT
Wolf Hirschhorn syndrome, also known as monosomy of the short arm of chromosome 4 (4p) or 4p-syndrome, is a rare genetic disorder first described in 1961 by doctors Cooper and Hirschhorn. The prevalence of this syndrome is extremely low, taking into account that the figure may be underestimated given the early gestational losses and the difficulty in prenatal diagnosis. The objective of the study is to present a clinical case of Wolf-Hirschhorn syndrome, presenting with multiple congenital morphological anomalies, as well as a neurological and intellectual retardation of variable degree. We report the case of a patient with a bicorial biamniotic twin gestation after a cycle of IVF-ICSI. The second twin was diagnosed with a Wolf-Hirschhorn syndrome, after performing the corresponding study due to a discordance of estimated weights and restricted intrauterine growth of this second fetus. The development of important craniofacial alterations, delay of normal prenatal and postnatal growth, and mental and intellectual deficiency of variable degree characterize the classic clinical presentation. Experts must make prenatal diagnosis. Wolf-Hirschhorn syndrome can be suspected by a restricted intrauterine growth, as it occurs in 80-90 % of fetuses with this pathology. Once diagnosed, the genetic study of the parents is recommended, since up to 15 % of the parents can suffer a balanced chromosomal rearrangement in the short arm of chromosome 4.
Keywords: Wolf-Hirschhorn syndrome; 4p deletion syndrome; congenital diseases; chromosomal alterations.
INTRODUCCIÓN
El síndrome de Wolf Hirschhorn (SWH), también conocido como monosomia del brazo corto del cromosoma 4 (4p) o síndrome 4p-,1,2 es una rara enfermedad genética descrita por primera vez en el año 1961 por los doctores Cooper y Hirschhorn.3-5 La prevalencia de esta patología es extremadamente baja. Aparece aproximadamente en 1 de cada 50 000 nacimientos, afecta dos veces más a mujeres que a hombres.1,2,4,6,7 Está causado por una delección pura en la porción 16 del brazo corto del cromosoma 4. No está asociado a otras anomalías citogenéticas que se produce en un 90 % de los casos como consecuencia de una mutación de novo.4,8 Sin embargo, existe un número pequeño de casos donde se pueden desarrollar alteraciones citogenéticas más graves, como el cromosoma 4 en anillo, los mosaicismos o translocaciones disbalanceadas.9,10
]]> Se destaca clínicamente por presentar un patrón específico de características craneofaciales que incluyen un puente nasal plano y ancho, frente alta, hipertelorismo y micrognatia, asociado a un retraso del crecimiento prenatal y posnatal, déficit intelectual y crisis epilépticas.11 El crecimiento intrauterino restringido fetal (CIR), aparece en torno al 80 % de los casos.1,2 El diagnóstico prenatal de esta anomalía a través del estudio morfológico mediante ecografía se puede considerar de alta complejidad. Objetivar las anomalías faciales típicas asociadas al crecimiento intrauterino restringido, debe alertar al ecografista para introducir esta infrecuente entidad dentro de las posibilidades diagnósticas.2El diagnóstico definitivo se establecerá mediante la realización de un estudio genético exhaustivo que incluya microarrays cromosómicos (CMA), análisis citogenético convencional con bandas G e hibridación in situ por fluorescencia (FISH).2 El tratamiento estará dirigido a las diversas manifestaciones que puedan aparecer. No existe un tratamiento único efectivo frente al síndrome.4 Dentro del diagnóstico diferencial de esta patología, el síndrome de Pitt Rogers Dank causado por una delección en el mismo locus genético, pero con una expresión clínica más atenuada, parece ser la principal entidad a tener en cuenta durante el estudio de este tipo de pacientes.6
El objetivo es presentar el caso de un feto diagnósticado de síndrome de Wolf Hirschhorn mediante el estudio de cariotipo con arrays cromosómicos tras amniocentésis, que debutó con un retraso de crecimiento intrauterino selectivo durante el seguimiento ecográfico de una gestación gemelar bicorial biamniótica.
PRESENTACIÓN DE CASO
Presentamos el caso de una mujer primigesta de 39 años de edad sin hábitos tóxicos y sin antecedentes de interés, estudiada en la Unidad de Medicina Reproductiva por esterilidad primaria de años de evolución. Tras someterse a técnicas de reproducción asistida mediante fecundación in vitro con inyección intracitoplasmática de espermatozoides (FIV-ICSI), la paciente consigue quedarse embarazada, manteniendo el tratamiento con yodo y ácido fólico para la prevención de alteraciones neuromedulares fetales. Tanto los estudios serológicos de toxoplasmosis, sífilis, hepatitis y VIH, como el cribado para aneuploidías cromosómicas realizados durante el primer trimestre de gestación, fueron negativos, en los que presentó inmunización para rubeola.
En el estudio ecográfico morfológico del primer trimestre, se confirmó la existencia de dos fetos anatómicamente normales con longitudes craneocaudales acordes con su tiempo de gestación y signo lambda interplacentario, que confirma el diagnóstico de embarazo gemelar bicorial biamniótico. Los controles posteriores se encuentran dentro de la normalidad, hasta que, a las 29 semanas de gestación, se objetiva en el estudio ecográfico una importante discordancia de pesos entre ambos fetos.
La paciente fue remitida a la Unidad de Diagnóstico Prenatal y Medicina Materno Fetal donde se visualizó una gestación gemelar bicorial biamniótica, con estudios morfológicos fetales y de volumen de líquido amniótico normales. Los índices de pulsatilidad (IP) y velocimetrías de flujo del estudio Doppler de la arteria umbilical, arteria cerebral media y ductus venoso de ambos fetos se encuentran dentro de la normalidad. Sin embargo, en las biometrías fetales se observó una importante discordancia de pesos estimados, el primer feto tuvo un peso de 1020 gramos (percentil 23), mientras que el segundo gemelo presentó un peso de 805 gramos (percentil 2), por lo que se confirmó el diagnóstico de crecimiento intrauterino restringido (CIR) selectivo del segundo feto en el contexto de una gestación gemelar bicorial biamniótica.
El comité multidisciplinar de la unidad decidió, tras la valoración del caso, realizar una amniocentésis para estudiar el cariotipo de ambos fetos, decisión que la paciente aceptó tras ser informada sobre los riesgos y beneficios de la técnica.
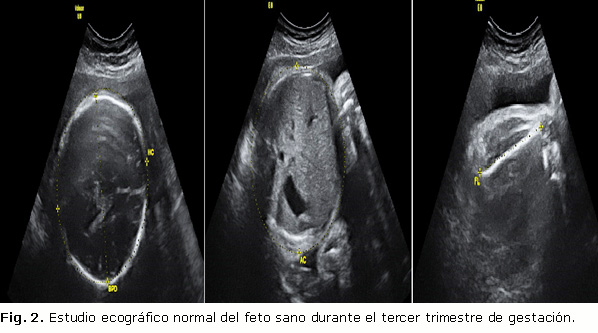
El Servicio de Genética del centro, tras estudiar los cromosomas de ambos fetos mediante la realización de cariotipo (Fig. 1) y arrays cromosómicos, evidenció las alteraciones cromosómicas compatibles con Síndrome de Wolf Hirschhorn en el feto que presentó un CIR selectivo (pérdida de 99Kb de ADN en la región cromosómica 4p16.3), mientras que el estudio del primer feto, cuyo peso se estima en un percentil 23, está dentro de la normalidad.
]]>
Tras el asesoramiento multidisciplinar, los progenitores se decidieron por la interrupción legal de la gestación del feto afectado. La paciente fue sometida a punción transdérmica guiada por ecografía. Se realizó la instilación a nivel intracardiaco fetal de 4 cm3 de cloruro potásico. Esto provocó la asistolia del gemelo afecto del SWH, confirmado mediante ecografía. Los controles ecográficos y cardiotocográficos posprocedimiento y de la gestación, evolucionan dentro de la normalidad (Fig. 2). A las 41+1 semanas de gestación, la paciente es derivada al servicio de obstetricia para inducción de parto por riesgo de embarazo prolongado y gestación gemelar bicorial-biamniótica con fetolisis selectiva del segundo gemelo. Tras el parto instrumental con ventosa obstétrica para abreviar expulsivo del primer gemelo, nace feto varón de 2b500 gr de peso y prueba de APGAR 9-10-10. El segundo feto de sexo femenino y peso de 780 gramos, nace posteriormente mediante parto eutócico con APGAR 0-0-0. Los controles puerperales de la paciente fueron normales. Recibió el alta hospitalaria dos días después del parto.
DISCUSIÓN
El síndrome de Wolf-Hirschhorn es un trastorno genético raro, infrecuente y sumamente desconocido, que cursa con múltiples anomalías morfológicas congénitas, así como con un retraso neurológico e intelectual de grado variable.4
La prevalencia total de esta enfermedad se estima en 1 por cada 50 000 recién nacidos. No obstante, esta cifra se puede considerar infraestimada por la dificultad diagnóstica de esta entidad, además de por las pérdidas gestacionales tempranas a las que se asocia y que son muy características de este síndrome. 9 La ratio de afectación mujer/hombre no es equitativa, ya que parece desarrollarse casi en el doble de los casos en el sexo femenino.1,2,4,6,7 En nuestro caso, la paciente presentaba una gestación gemelar bicorial biamniótica con fetos de distinto sexo, siendo el gemelo de sexo femenino el afectado por esta patología. La variabilidad en el espectro de manifestaciones clínicas del Síndrome Wolf-Hirschhorn ha sido atribuida al tamaño del defecto cromosómico relacionado con la delección genética, producida en la región critica 16,3 del brazo corto del cromosoma 4, confinada en 165 kilobases.4,10
A pesar de que la presencia única de microdelecciones en la porción 16 del brazo corto del cromosoma 4 (con ocupación menor de 5 megabases), está relacionada con las manifestaciones clínicas más características de esta patología, constituyen menos del 3 % de todos los casos reportados en la literatura.2 Sin embargo, la mayoría de los pacientes afectados por esta rara entidad, presentan una delección de mayor tamaño que la anteriormente descrita.2 El patrón clásico de presentación clínica se caracteriza por el desarrollo de alteraciones craneofaciales importantes, retraso en el crecimiento normal tanto prenatal como posnatal y deficiencia mental e intelectual de grado variable.11 La alteración craneofacial más característica es el puente nasal ancho y ampliado, que se continua hasta la frente y confiere a estos pacientes una apariencia denominada en "casco de guerrero griego".4,6,8
Esta manifestación es fácilmente reconocible desde el nacimiento y durante la infancia, pero se vuelve menos evidente con la edad, puede ser casi imperceptible en la pubertad.2 La presencia de otras alteraciones craneofaciales como frente alta, hipertelorismo, cejas arqueadas y elevadas, epicantus, microcefalia y micrognatia se desarrollan con menor frecuencia y dependen del defecto genético asociado al tamaño de la delección cromosómica.2,6,8
El crecimiento intrauterino restringido se presenta en 80 % de los casos,1,2 lo que constituye el hallazgo ultrasonográfico prenatal más frecuentemente detectado, por lo que puede estar asociado a otras anomalías fetales.9,12
En nuestro caso, la paciente fue diagnosticada en el tercer trimestre de gestación de un CIR selectivo en unos de los gemelos, lo que derivó en los posteriores estudios genéticos que llevaron al diagnóstico definitivo del síndrome. Todos los pacientes que presentan esta alteración genética desarrollarán retraso intelectual y mental en grado variable, variabilidad que dependerá del tamaño de la afectación cromosómica que presenten.8,13 Otras manifestaciones clínicas como la hipotonía, anomalías cardiacas y esqueléticas, defectos en el nervio óptico o en el tracto genitourinario pueden estar presentes, aunque la frecuencia de aparición de estas, es mucho menor.1,2
]]> El diagnóstico de sospecha prenatal se basa en el estudio ecográfico morfológico fetal, aunque posteriormente requiere la confirmación del mismo mediante el estudio genético de las células fetales.9 En la mayoría de los estudios publicados en la literatura, se ha obtenido un diagnóstico definitivo mediante la aplicación de análisis citogenético convencional tras amnio/cordocentésis o biopsia corial, pero en la última década, las nuevas tecnologías como los microarrays han permitido una descripción más precisa y exhaustiva de los mecanismos moleculares que definen el fenotipo característico del Síndrome Wolf-Hirschhorn.7,9 De hecho, existen artículos publicados de casos clínicos en los que, tras la interpretación de un cariotipo normal por parte de los genetistas, se identificaron alteraciones moleculares posteriores mediante técnicas como microarrays o FISH, que habían pasado desapercibidas en los estudios previos.9El análisis citogenético convencional puede detectar entre 50 y 60 % de las alteraciones genéticas características de este síndrome, mientras que con la aplicación de la técnica FISH se consigue detectar más del 95 % de las delecciones cromosómicas de este.9
El estudio mediante cariotipo molecular o microarrays es la técnica de elección en la actualidad, ya que puede detectar valores cercanos al 100 % de las delecciones conocidas actualmente y, además determinar si está es "pura" o parte de un desequilibrio más complejo, con una mayor precisión que con las dos técnicas anteriormente mencionadas.
En la actualidad, se recomienda la realización de técnicas de alta resolución como los microarrays o FISH, ante una discordancia en los hallazgos encontrados entre el estudio sonográfico prenatal patológico y el resultado normal del cariotipo, obtenido mediante estudio citogenético convencional.9,13 En nuestro caso se realizó una amniocentesis en el tercer trimestre de gestación, para estudiar el material genético mediante microarrays. Se obtuvo como resultado alteraciones genéticas del feto con CIR selectivo, compatibles con Síndrome de Wolf-Hirschhorn.
Se estima que en 85-90 % de los casos, esta anomalía cromosómica congénita, no han sido heredada, ya que suelen desarrollarse a partir de mutaciones de novo. Sin embargo, en 10-15 % de los pacientes afectos, uno de los progenitores presentará un reordenamiento cromosómico equilibrado en la región 16,3 del brazo corto del cromosoma 4. 4 Por esta razón, se recomienda el análisis cromosómico y estudio genético de los progenitores, así se podrán determinar los futuros riesgos de desarrollar patologías genéticas y recibir un asesoramiento genético correcto, permitiendo una planificación adecuada y consciente de sus deseos genésicos.4
El pronóstico a largo plazo de este tipo de síndromes, estará directamente relacionado con las diferentes manifestaciones clínicas que se presenten en el paciente. En los últimos años, el mejor conocimiento de la enfermedad, así como la mejora en la atención sanitaria y social, han contribuido al aumento de la supervivencia a largo plazo.4 Algunos estudios refieren tasas de mortalidad del 34 % dentro de los dos primeros años de vida, aunque se ha descrito casos de supervivencia hasta los 30 años de edad.4,14
CONCLUSIONES
El síndrome de Wolf-Hirschhorn es un síndrome genético extremadamente raro y desconocido, causado por una mutación en el brazo corto del cromosoma 4, en la mayoría de los casos, producido de novo. La incidencia es muy baja y difícil de determinar con exactitud, ya que se trata de un diagnóstico complejo desde el punto de vista prenatal, caracterizado ecográficamente por el crecimiento intrauterino restringido. La triada clásica que se destaca en esta patología son las alteraciones craneofaciales, retraso en el crecimiento normal tanto prenatal como postnatal y deficiencia mental e intelectual de grado variable. El pronóstico a largo plazo es inestable, el diagnóstico prenatal es de vital importancia, realizado por ecografistas expertos. Destacar la necesidad del asesoramiento genético en los progenitores ya que existen alrededor de un 10-15 % de los casos en los que la transmisión es hereditaria.
]]> Conflictos de intereses
REFERENCIAS BIBLIOGRÁFICAS
1. Battaglia A, Filippi T, Carey JC. Update on the clinical features and natural history of Wolf-Hirschhorn (4p-) syndrome: experience with 87 patients and recommendations for routine health supervision. Am J Med Genet Part C Semin Med Genet. 2008;148C(4):246-51.
2. Battaglia A, Carey JC, South ST. Wolf-Hirschhorn Syndrome: A Review and Update. American Journal of Medical Genetics Part C (Seminars in Medical Genetics). 2015;169C:216-23.
3. Bergemann AD, Cole F, Hirschhorn K. The etiology of Wolf-Hirschhorn syndrome. Trends Genet. 2005;21(3):188-95.
4. Bailey R. Wolf-Hirschhorn Syndrome. A Case Study and Disease Overview. Advances in Neonatal Care. 2014.318-321
5. Copper HL and Hirschhorn K. Apparent deletion of short arms one chromosome (4 or 5) in a child with defect of midline fusion. Chromosome Newslett. 1961;IV(14).
6. Ikonomou T, Panos A, Daskalakis G, Sindos M, Papantoniou N, Kosmaidou Z. Prenatal diagnosis of Wolf- Hirschhorn syndrome: ultrasonography and genetics. J Matern Fetal Neonatal Med. 2013;26(9):941-2.
7. Saberi A, Shariati G, Hamid M, Galehdari H and Abdorasouli N. Wolf-Hirschhorn Syndrome: A Case with Normal Karyotype, Demonstrated by Array CGH (aCGH). Archives of Iranian Medicine. 2014;(17)9.
8. Genetic Home Reference. Wolf-Hirschhorn syndrome. 2009. Disponible en: http://ghr.nlm.nih.gov/condition=wolfhirschhornsyndrome#inheritance
9. Sifakis S, Manolakos E, Vetro A, Kappou D, Peitsidis P, Kontodiou M, et al. Prenatal diagnosis of Wolf-Hirschhorn syndrome confirmed by comparative genomic hybridization array: report of two cases and review of the literature. Mol Cytogenet. 2012;5:12.
10. Paradowska-Stolarz AM. Wolf-Hirschhorn Syndrome (WHS)- Literature Review on the Features of the Syndrome. Adv Clin Exp Med. 2014;23(3):485-9.
11. Ho KS, South ST, Lortz A, Hensel CH, Sdano MR, Vanzo RJ et al. Chromosomal microarray testing identifies a 4p terminal region associated with seizures in Wolf-Hirschhorn syndrome. J Med Genet. 2016;53(4):256-63.
12. Ferreres García K, Viñuela Benítez MC, Cervantes Reyna D, Blanco Soto P, Gámez Alderete F. Retraso del crecimiento intrauterino de causa genética: síndrome de Wolf-Hirschhorn. Prog Obstet Ginecol. 2017;60(1):75-8.
13. Hajdu I, Ciccia A, Lewis S, Elledge S. Wolf-Hirschhorn syndrome candidate 1 is involved in the cellular response to DNA damage. Proc National Acad Sci. 2011;108(32):13130-4.
14. Lopes O, Barton G, Morgan J. Wolf-Hirschhorn syndrome-two cases study reports focusing on particularly on long-term survival. J Intellect Disabil Res. 2005;49(3):228-30.
Recibido: 14 de agosto de 2017.
Aprobado: 3 de septiembre de 2017.
Manuel Pantoja Garrido. Unidad de Gestión Clínica de Obstetricia y Ginecología del Hospital Universitario Virgen Macarena de Sevilla, España.
Correo electrónico: pantoja_manuel@hotmail.com
]]>