Esferocitosis hereditaria: de la biogénesis a la patogénesis
Hereditary spherocytosis: from biogenesis to pathogenesis
]]>
Dra. Heidys Garrote-Santana,I Lic. Michel Gómez-Pacheco,II Dr. Juan Carlos Jaime-Fagundo,I Dra. Valia Pavón-Morán,I Dra.Cs. Gisela Martínez-AntuñaI
I Instituto de Hematología e Inmunología. La Habana, Cuba.
II Instituto de Ciencias Básicas y Preclínicas "Victoria de Girón". La Habana, Cuba.
RESUMEN ]]>
La esferocitosis hereditaria es la anemia hemolítica congénita más frecuente en la población caucásica. Tiene una amplia variabilidad clínica y desde el punto de vista hematológico se caracteriza por anemia y presencia de esferocitos en la lámina periférica. Su base fisiopatológica está determinada por el defecto de algunas de las proteínas que conforman la membrana eritrocitaria, por el efecto del bazo sobre los hematíes anómalos y otros factores. A la luz de los conocimientos actuales, la interpretación dinámica del proceso requiere adentrarse en los estadios iniciales de la hematopoyesis, pues desde etapas tan tempranas como la enucleación del eritroblasto en la formación del reticulocito, hasta posibles procesos inflamatorios tardíos, pudieran modular la expresión de la enfermedad. Se hace una revisión de las características estructurales y funcionales de la membrana eritrocitaria, así como algunos aspectos generales de las propiedades del hematíe para facilitar la comprensión de los eventos que tienen lugar a partir del compromiso molecular de las proteínas que conforman la membrana.
Palabras clave: esferocitosis hereditaria, membrana eritrocitaria, proteínas de membrana.
ABSTRACT
Hereditary spherocytosis is the most common congenital hemolytic anemia among Caucasian population. It has wide clinical variety and from the haematological point of view, it is characterized by the presence of spherocytes anemia in peripheral lamina. Its pathophysiological defect is determined by some of the proteins that make up the red cell membrane due to the effect on erythrocytes of abnormal spleen, and other factors. In view of current knowledge, the dynamic interpretation of this process requires delving into the early stages of hematopoiesis, since the expression of this disease could modulate from early stages of erythroblast enucleation in reticulocyte formation until late potential inflammatory processes. A review was made on the structural and functional characteristics of the erythrocyte membrane, as well as some general aspects of the properties of the red cell to facilitate understanding of events which take place through proteins molecular involvement forming the membrane.
Keywords: hereditary spherocytosis, erythrocyte membrane, membrane proteins.
]]>
INTRODUCCIÓN
La esferocitosis hereditaria (EH) es un trastorno hemolítico que se produce por la deficiencia de algunas de las proteínas que conforman la membrana eritrocitaria. Se caracteriza por la presencia de esferocitos en la lámina periférica, anemia, ictericia intermitente, esplenomegalia y en la mayoría de los casos severos, buena capacidad de respuesta a la esplenectomía.1,2
Para comprender la participación de cada gen o proteína en la patogénesis de la EH es necesario mirar en la biogénesis de la membrana durante la eritropoyesis.
Debido a su fácil accesibilidad, la membrana eritrocitaria es la más estudiada de las membranas biológicas.1 Compuesta por una bicapa lipídica (los lípidos son del 50-60 % de la masa de la membrana) y un esqueleto subyacente, provee al eritrocito de la estabilidad y deformabilidad requeridas para viajar a través de la circulación.2
]]>
BICAPA LIPÍDICA
Está constituida fundamentalmente por fosfolípidos y colesterol, con pequeñas cantidades de glicolípidos. Los fosfolípidos primarios son: fosfatidilcolina (28 % del total de fosfolípidos), fosfatidiletanolamina (27 %), esfingomielina (26 %), fosfatidilserina (13 %) y en menor cuantía, fosfatidilinositol.2 Estos fosfolípidos están distribuidos de forma asimétrica, lo que parece ser importante para mantener la homeostasis. Los glicolípidos y el colesterol están intercalados entre los fosfolípidos.
Los lípidos están organizados en 3 dominios dentro de la bicapa, los que se dividen en: dominios macroscópicos rico en lípidos, dominios microscópicos unidos a proteínas, y dominios asociados con la fracción detergente-resistente de la membrana (DRM) o balsas de lípidos.1,3,4 La función de estos dominios aún no está clara, pero se supone que los DRM pueden facilitar la penetración de la malaria en el eritrocito,4,5 para adicionarse a los múltiples eventos que se describen en la fisiopatología de esta entidad.6
PROTEÍNAS ]]>
La membrana del eritrocito contiene alrededor de 15 proteínas mayores y cientos de menores. En estudios proteómicos del eritrocito se ha encontrado que de 527 proteínas alrededor de 340 están asociadas con la membrana.7,8
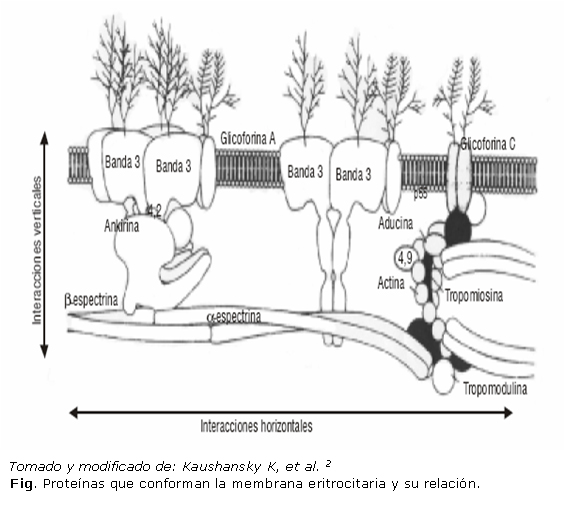
Tradicionalmente, estas proteínas se clasifican de acuerdo con la facilidad con la que pueden ser eliminadas totalmente de la membrana.2 Proteínas integrales: son aquellas que están arraigadas firmemente y distribuidas a través de la bicapa lipídica e interactúan con los lípidos hidrofóbicos (glicoforinas, las proteínas Rh, los antígenos Kell y Duffy, proteínas transportadoras como la banda 3); las proteínas periféricas que están asociadas de manera más lábil, por lo que su extracción es más fácil. Estas interactúan (por uniones covalentes y no covalentes) con las proteínas integrales o los lípidos que recubren la membrana, pero no penetran dentro de la bicapa y solo se relacionan con la cara citoplasmática (proteínas estructurales del esqueleto: espectrinas, ankirina, actina, proteína 4.1 y proteína 4.2).1 (Fig.)
Estas proteínas se han nombrado de acuerdo con la movilidad electroforética en gel de poliacrilamida-dodecil sulfato, en base al orden de migración de la banda de la proteína.9 Algunas de ellas aún se nombran con esos términos, como la banda 3 o las proteínas 4.1 y 4.2.
]]>
Proteínas integrales
Banda 3 (intercambiador de aniones):
Proteína de abundante distribución que funciona como regulador del contenido de iones; es intermediaria del metabolismo del hematíe y contribuye a la deformabilidad del glóbulo rojo. Además, posiblemente interviene en el proceso de la senescencia del eritrocito.2,10,11
Es la mayor intercambiadora de aniones (cloro-bicarbonato) en el hematíe. Su participación en el metabolismo intraeritrocitario está relacionada con vías metabólicas donde intervienen enzimas glicolíticas como la gliceraldehido 3 fosfato deshidrogenasa, la fosfoglicerato cinasa, y la aldolasa, así como las anhidrasas carbónicas II y IV.2,5
La banda 3 contiene importantes sitios de unión para interactuar con otras proteínas de la membrana, tales como la ankirina, la proteína 4.1 y la proteína 4.2. La unión del dominio citoplasmático con la ankirina es uno de los mecanismos críticos para anexar el esqueleto a la membrana plasmática, lo que es determinante en la flexibilidad o rigidez del eritrocito.11 Los dominios extracelulares de la banda 3 constituyen antígenos para diferentes grupos sanguíneos.10
]]>
Proteínas periféricas
Espectrina
Es la proteína más abundante y larga de las que conforman el esqueleto de membrana. Está formada por subunidades a y b, que aunque tienen semejanzas, son estructuralmente diferentes y están codificadas por genes separados.1,2
Ambas subunidades contienen 106 aminoácidos homólogos que están situados en segmentos helicoidales. La presencia de estas repeticiones sugiere que las 2 cadenas de la espectrina se desarrollaron a partir de la duplicación de un solo gen ancestral.2
La estructura de triple hélice de la molécula de espectrina está dada por heterodímeros a b, que se alínean y entrelazan entre sí de modo antiparalelo con respecto a sus terminales NH2, para formar heterotetrámeros.
Estos tetrámeros compuestos de las repeticiones múltiples, suministran un filamento fuerte y elástico que interviene de manera decisiva en la forma y resistencia de la membrana. Además, la espectrina regula la movilidad lateral de las proteínas de membrana, garantiza el soporte estructural para la bicapa lipídica y se une a la actina y a la proteína 4.1 mediante secuencias repetitivas.2 Otras secuencias no repetitivas en la espectrina proveen de sitios para el reconocimiento y unión de otros moduladores como cinasas y calmodulina.2
Aunque los miembros de la familia de las espectrinas adoptan un pliegue helicoidal repetitivo, las interacciones muy específicas de las espectrinas con otras proteínas indican que las repeticiones han evolucionado para proveer una función especializada mientras se mantiene la estructura terciaria.12
Ankirina
En los humanos existen 3 isoformas: ankirina 1 o R, que se encuentra en el eritrocito; ankirina 2 o B, localizada primariamente en el cerebro; y la ankirina 3 o G, con una expresión más general. ]]>
Esta proteína puede ser separada en 3 dominios funcionales: uno NH2 terminal de unión a la membrana que contiene sitios para la banda 3 y otros ligandos; un dominio central que contiene sitios para las espectrinas; y uno COOH terminal regulador, que influye en la interacción con otras proteínas.12,13El dominio de unión a la membrana contiene 24 repeticiones en tándem, las que contienen múltiples sitios de unión con proteínas. Las repeticiones de la ankirina son altamente conservadas y se han encontrado en otras proteínas con gran variedad de funciones.2,12
La ankirina garantiza la unión al esqueleto de membrana mediante su relación con la espectrina y a la bicapa lipídica a través de la banda 3. Las interrupciones de alguna de estas vías de unión provocan inestabilidad en la membrana.
Proteína 4.2
Pertenece a la familia de las transglutaminasas;14 sin embargo, la proteína 4.2 no posee actividad de transglutaminasa porque carece de un residuo crítico en su sitio activo. Se han descrito al menos 4 isoformas de la proteína, aunque la trascendencia funcional de las isoformas no está esclarecida. La proteína 4.2 une diferentes proteínas: la banda 3, la proteína 4.1 y la ankirina. Su principal función es estabilizar la asociación del complejo espectrina-actina-ankirina con la banda 3.2,15 También protege el esqueleto de la membrana del envejecimiento prematuro mediante la unión del calcio y otros cofactores que normalmente activan las transglutaminasas en el hematíe.
]]>
ESTRUCTURA Y FUNCIÓN DE LA MEMBRANA
La función de la membrana eritrocitaria incluye el ensamblaje y organización de las proteínas de la bicapa lipídica con las del esqueleto, proporciona a la célula la estabilidad y deformabilidad necesarias, participa en la biogénesis y el envejecimiento, y provee de una barrera entre el citoplasma y el medio externo con una permeabilidad selectiva.
Ensamblaje y organización
]]>
Cuando se examina la membrana eritrocitaria y esta es estirada, las proteínas individuales del esqueleto aparecen como un enrejado organizado de hexágonos. Las esquinas de cada hexágono son estructuras globulares llamadas complejos de unión formados por actina, aducina y proteína 4.1. Los tetrámeros de espectrinas forman los brazos de los hexágonos. Estos tetrámeros, en sus colas, se unen con los complejos de unión de actina con la ayuda de la proteína 4.1 y la aducina. Las interacciones horizontales (paralelas en el plano de la membrana) derivadas de las uniones anteriores son vitales para el mantenimiento de la integridad estructural de la célula.2,5,6 (Fig.).El esqueleto se fija a las proteínas integrales de la membrana por varias interacciones proteicas. La espectrina es el componente principal de la red que está anclada a la membrana por la ankirina (proteína adaptadora), que a su vez establece el vínculo de la espectrina con la banda 3.12,16
Los extremos terminales de las espectrinas también se unen con la membrana mediante la proteína 4.1, la cual se fija a la glicoforina C y la P55. Adicionalmente, la espectrina y la proteína 4.1 se unen débilmente con la fosfatidilserina ubicada en el interior de la bicapa lipídica.2,5 Estas interacciones verticales (perpendiculares en el plano de la membrana): proteína-proteína y proteína-lípido, son críticas en la estabilidad de la bicapa lipídica e impiden la pérdida de sus celdas (Fig.).
Estabilidad de la membrana y deformabilidad celular
La deformabilidad es la propiedad más importante del hematíe para su supervivencia y donde mejor se pone a prueba es en su tránsito por los senos esplénicos.17 Se describen 3 factores determinantes en la deformabilidad: la geometría celular (forma de disco bicóncavo), la viscosidad citoplasmática (determinada por la concentración de hemoglobina en la célula) y la elasticidad intrínseca de la membrana (deformabilidad de la membrana).2,5,18 De estos, la geometría celular se considera el factor más importante. ]]>
El ser un disco bicóncavo le proporciona al hematíe una alta relación superficie/volumen (140 µm3/90 µm3); necesaria para diferentes situaciones, como cuando el hematíe se agranda por estrés osmótico o cuando se estira bajo los efectos del estrés mecánico de la circulación.2,5,6La organización del esqueleto y su anexo con la membrana también influye en la estabilidad y deformabilidad del hematíe. La alta elasticidad de la membrana es determinada en forma general por su estructura en red y en particular, por la espectrina. En reposo los segmentos helicoidales de espectrina están altamente enrollados. Cuando la membrana se deforma se establece una restructuración de la red espectrina-actina: algunas moléculas de espectrina comienzan a desenrollarse y extenderse, mientras que otras se comprimen y mantienen sin variación el área de superficie.2,5
La viscosidad del hematíe es determinada en gran parte por los niveles de hemoglobina. A las concentraciones intracelulares normales de hemoglobina la viscosidad tiene muy poca contribución en la deformabilidad.
La concentración de hemoglobina corpuscular media en la célula humana normal es de alrededor de 33 g/dL. Mientras la viscosidad de la hemoglobina es solo 5 centipoises (cp) a los 27 g/ dL, se incrementa a 15 cp a los 37 g/dL; a 45 cp a los 40 g/dL; a 170 cp a los 45 g/dL; y a 650 cp a los 50 g/dL.5 Al regular la concentración de Hb dentro de este estrecho margen, se minimizan los efectos de la viscosidad citoplasmática en la deformabilidad del hematíe.
La habilidad de la célula para acomodarse rápidamente en un tubo capilar angosto en la microcirculación, se verá comprometida por el incremento de la viscosidad citoplasmática. Es importante señalar que la deshidratación del hematíe y el aumento de la viscosidad citoplasmática comprometen la supervivencia del glóbulo rojo en grado mínimo.5
Otros estudios destacan 3 importantes factores que influyen en la estructura e integridad mecánica de la membrana, así como el mantenimiento de la forma del hematíe: 1) el cizallamiento sobre la membrana; 2) la naturaleza de las uniones verticales y su pérdida; 3) el remodelado facilitado por la energía derivada de la actividad metabólica.6
Algunos sitios del citoesqueleto de la membrana, como la red de espectrina, pueden absorber energía química proporcionada por moléculas como el ATP. El número de eventos de transferencia molecular de energía ocurridos por unidad de tiempo es llamado hit de energía. La red de espectrina se fluidiza mientras mayor es el hit de energía. El cizallamiento influye en la fluidización o plasticidad de la red de espectrina. Cambios en los niveles de ATP en condiciones experimentales inducen a cambios en la forma del eritrocito e incremento de las fluctuaciones de la membrana. En vivo, las modificaciones de la energía en la membrana, secundarias al cizallamiento (infecciones, traumas, trastornos hereditarios, envejecimiento celular, cáncer), determinan sus fluctuaciones. La unión de la bicapa lipídica y la red de espectrina es crítica y está controlada por los cambios del ATP.6
]]>
Propiedades de la membrana
Las propiedades de la membrana son un reflejo de las características de la bicapa lipídica y el esqueleto. Durante la deformación se describe un rápido traslado del colesterol del interior al exterior de la bicapa. Los hematíes que están suspendidos en soluciones hipotónicas, como las que se utilizan en el estudio de fragilidad osmótica, se agrandan y adoptan una forma esférica ya que la bicapa de la membrana no puede expandir su área de superficie más allá del 3 o el 4 %. El descenso de la presión osmótica resulta en la ruptura de la membrana y la hemoglobina intracelular es descargada al exterior.2
La membrana del hematíe es casi impermeable a los cationes mono y divalentes; de esta forma, mantiene alta la concentración de potasio intracelular, baja la de sodio y muy bajos los niveles de calcio. Por el contrario, es muy permeable al agua y a los aniones que son intercambiados fácilmente, razón por la que se plantea que el hematíe funciona como un osmómetro perfecto. La glucosa es transportada sin consumo de energía.
Las vías para el transporte de agua e iones en el eritrocito incluyen: bombas de membrana dependientes de energía, sistema de gradientes y varios canales.
Bombas de membrana dependientes de energía: ]]>
La bomba Na+-K+ ATPasa es la encargada de mantener el sodio intracelular bajo y el potasio alto, con una relación de intercambio 3:2. Tiene una importante función en el mantenimiento del pH citosólico, la homeostasis iónica y la osmolaridad celular.19 La Ca2 + ATPasa es activada por la calmodulina, extrae calcio de la célula y así mantiene concentraciones intracelulares muy bajas, lo que la protege de los múltiples efectos deletéreos del calcio: equinocitosis, vesiculación de la membrana, proteólisis y deshidratación celular. La membrana también contiene un transportador ATP de glutatión oxidado y un sistema transportador de aminoácidos.2,5
Sistema de gradientes:
El gradiente de Na+-K+ establecido por la bomba, es usado por múltiples sistemas de gradientes pasivos para mover iones a través de la membrana. Estos sistemas incluyen: el cotransportador K+-Cl-, la banda 3, el cotransportador Na+-K+-Cl- (de importancia discreta dentro de la célula) y el intercambiador de Na+-H+ (parece ser protagónico en la maduración temprana del hematíe). El cotransportador K+-Cl- es activo en los reticulocitos20 y su actividad se acelera por la inflamación, acidificación y depleción del magnesio intracelular.2,5,21
Canales:
Los canales mediados por Na+-K+ ATPasa, canales de agua (acuaporinas) y los canales de K+ activados por Ca2+ (canal Gardos), causan la pérdida selectiva de K+ en respuesta al incremento intracelular de Ca2+.2
Las acuaporinas son canales de proteínas que sirven como poros selectivos a través de los que pasa el agua y contribuyen a la habilidad del eritrocito para hacer ajustes rápidos ante los cambios de la osmolaridad.22,23
Se ha demostrado por monitoreo simultáneo del pH y marcadores de volumen, un enlace funcional entre el transporte de iones de la membrana, el pH y el volumen celular;24 la hidratación adecuada es un factor de peso para evitar la formación de vesículas en la superficie de la membrana.25
]]>
ESFEROCITOSIS HEREDITARIA
Genética
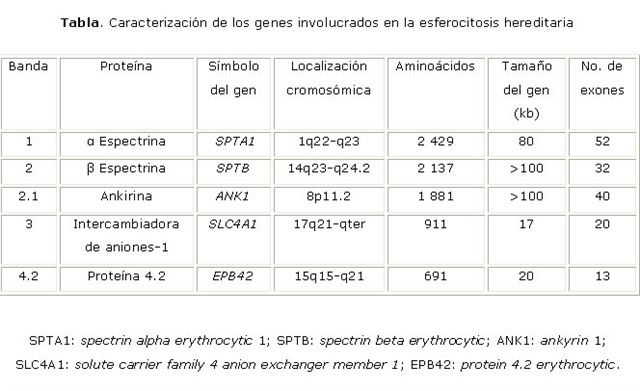
Se han caracterizado los genes que codifican para las diferentes proteínas de la membrana eritrocitaria y en particular aquellas involucradas en la esferocitosis (tabla).
En la mayoría de familias afectadas, la EH es transmitida de manera autosómica dominante (75 %) con un padre enfermo. El 25 % expresa un patrón autosómico recesivo según reportes de familias con padres aparentemente normales y más de un hijo afectado.1 ]]>
Las mutaciones responsables de la EH incluyen los genes de las ? y ? espectrinas (SPTA 1 y SPTB, respectivamente), la ankirina (ANK1), la banda 3 (SLC4A1) y la proteína 4.2 (EPB42).6 Se han descrito mutaciones espontáneas o de novo, particularmente en los genes ANK1 y SPTB.1
Patogenia
La lesión primaria de la EH es causada por el defecto de las proteínas de membrana. El primer defecto bioquímico reconocido en los pacientes con esferocitosis fue la deficiencia de espectrina. Además, se describen: deficiencia combinada de espectrina y ankirina, deficiencia de banda 3, deficiencia aislada de espectrina, y deficiencia de 4.2.1,2,26,27
Espectrinas
En el eritrocito, las a-espectrinas se sintetizan 3-4 veces por encima de las b-espectrinas2,28 y el exceso de las primeras se degrada. Las anormalidades clínicas causadas por las deficiencias de estas cadenas se encuentran en los homocigóticos o dobles heterocigóticos. Los pacientes heterocigóticos producen suficientes cadenas a, para balancear con la producción de b-espectrinas y mantener un fenotipo normal. En contraste con lo anterior, las deficiencias de b-espectrina se hacen evidentes clínicamente en el heterocigótico por su velocidad de síntesis limitado.1 ]]>
a-espectrinaLas determinaciones de las alteraciones genéticas precisas asociadas con el gen de la a-espectrina han sido difíciles debido al tamaño del gen (52 exones), así como a la reducción del RNA mensajero, asociado con la presencia de alelos nulos.1
Se han descrito algunos alelos asociados con la forma recesiva de la EH. La a-espectrinaLEPRA es una sustitución de citosina por timina en el sitio e empalme del intrón 30 que crea un sitio de empalme alternativo.1-2 La combinación del alelo LEPRA con otro defecto de a-espectrina en trans, una cadena de a-espectrina truncada o una aPrague, resulta en una deficiencia severa de la proteína y por lo tanto, una anemia significativa.
La sustitución de un aminoácido en el dominio aII de la espectrina (969 GCT® GAT: Ala ® Asp) determina el alelo a-espectrinaBug Hill identificado en muchos pacientes con deficiencia recesiva de espectrinas.
b-espectrina
Se ha descrito un grupo de pacientes heterocigóticos para el defecto en las cadenas de las b-espectrinas. Estos pacientes padecen de EH típica2,29 con una subpoblación de acantocitos. La mayoría de las mutaciones de b-espectrina se asocian con alelos nulos e incluyen mutaciones sin sentido y mutaciones del codon iniciador.2 También se han descrito mutaciones espontáneas.
Las mutaciones de la b-espectrina afectan la síntesis o producen cadenas truncadas que son inestables o no se unen con la ankirina, lo que impide el ensamblaje al esqueleto de membrana.21 Una de las variantes descritas es la b-espectrinaKissimmee, que tiene una mutación puntual que produce inestabilidad de la cadena y un defecto en la unión de la espectrina con la proteína 4.1 con débil unión a la actina.2 La espectrinaHouston, se produce por deleción de un nucleótido.
Se han descrito cadenas truncadas, como es el caso de la espectrinaBari, una proteína producida por una mutación puntiforme en el sitio de empalme del intrón 16, que conlduce a la síntesis de un ARNM que tiene una deleción de los exones 16 y 17 de la cadena.30 Otras mutaciones del gen b-espectrina que han sido enunciadas recientemente son: R1756X, 781delT , IVS22nt-4G>A, 1502insA y IVS20nt-2A>G.31
Ankirina ]]>
Se han propuesto varios mecanismos relacionados con el compromiso de la ankirina: disminución en la síntesis, disminución en el ensamblaje a la membrana y ensamblaje de una proteína anormal.Los estudios genéticos han identificado un número de mutaciones en el gen de la ankirina y se ha demostrado que los defectos de esta proteína son la causa más común de EH típica dominante. La mayoría de estas mutaciones son sin sentido, que resultan en un defecto de la molécula de ankirina, una deficiencia de la proteína o ambas.2 También se han descrito mutaciones de novo como E9X (p.Glu9Term, c.25G>T, GAA>TAA) en el exón 1.32
La combinación de deficiencia de espectrina y ankirina es la anomalía bioquímica más frecuente en los pacientes con EH típica dominante. La ankirina es el sitio principal de unión de la espectrina, por lo que la deficiencia de ankirina disminuye la incorporación de la espectrina a la membrana, a pesar de que la espectrina se sintetice de manera adecuada.1
Algunas de las variantes descritas son: la ankirinaWalsrode, cuya mutación afecta el dominio de unión a la banda 3 y por lo tanto, disminuye la afinidad por esta; y la ankirinaFlorianópolis, que está asociada con la EH dominante.
En algunos pacientes con EH recesiva hay variantes identificadas con alteraciones en la región promotora del gen de la ankirina.2 Se han descrito alrededor de 55 mutaciones diferentes en el gen que codifica para esta proteína.32
Estudios experimentales describen posibles mutaciones nuevas como es el caso de Ank1E924X, con compromiso de los sitios de unión con la espectrina.33
Investigaciones recientes exponen la participación del gen de la ankirina en mecanismos de regulación genética, que funciona como barrera de aislamiento entre diferentes dominios cromáticos.34 Algunos estudios citogenéticos han identificado pacientes deficientes de ankirina con alteraciones dismórficas, retraso psicomotor e hipogonadismo. Estos pacientes sufren de un síndrome genético que compromete el gen de la ankirina: 8p11.2.2
Banda 3
Se ha planteado que existe un grupo de pacientes (10-25 %)1,2 con EH dominante típica, en los que las células son deficientes en el 20-40 % para la proteína banda 3 y tienen además una disminución o ausencia de la 4.2, con un contenido normal de espectrinas. Estos pacientes generalmente tienen una anemia moderada y hematíes del tipo pincered cell (apariencia de hongo) en la lámina periférica. El transporte de aniones está comprometido proporcionalmente con la deficiencia de la proteína.1 ]]>
Han sido enunciadas una variedad de mutaciones en el gen que codifica para la banda 3, tales como inserciones, deleciones, mutaciones puntuales, sin sentido y mutaciones en el procesamiento del ARN.35Algunas mutaciones remplazan residuos de arginina muy conservados en el dominio transmembrana. Las proteínas mutadas no se pliegan adecuadamente, lo que compromete sus interacciones o afecta su tráfico celular del retículo endoplasmático a la membrana eritrocitaria.1 Las mutaciones sin sentido resultan en la disminución de la acumulación de ARNm presumiblemente por inestabilidad.
En la EH con banda 3Campinas y banda 3Pribram, que provocan alteraciones en el procesamiento del ARNm, se ha descrito una acidosis tubular asociada.2 Esto pudiera estar relacionado con la expresión de la banda 3 en los túbulos distales del riñón. Sin embargo, solo un número reducido de pacientes con EH se asocia con defectos en la acidificación urinaria.36-38 Estudios recientes describen la mutación banda 3Edmonton 1 también asociada con acidosis tubular renal.39
Proteína 4.2
La deficiencia de la proteína 4.2 con patrón de herencia recesivo, fue descrita inicialmente en pacientes japoneses en los que se demostró una marcada reducción de la proteína.38,40 La variante 4.2Nippon es causada por una mutación puntiforme que presumiblemente afecta el procesamiento del ARNm.41 Otras variantes han sido descritas en pacientes homocigóticos o dobles heterocigóticos. Estas deficiencias también han sido observadas en pacientes con mutaciones en el dominio citoplasmático de la banda 3, las que presumiblemente involucran la región de la banda 3 que interacciona con la 4.2.1,2,42,43
DEFECTOS SECUNDARIOS EN LA MEMBRANA ]]>
Permeabilidad de la membrana y contenido de cationes
En la EH, el contenido de agua y potasio en los hematíes está disminuido. La permeabilidad pasiva para el sodio está incrementada, posiblemente secundario al defecto del esqueleto subyacente. El influjo excesivo del sodio activa la bomba Na+-K+ ATPasa, lo que acelera la entrega de ATP y la glicolisis.2
Las vías que causan deshidratación de los hematíes en la EH aún no están claras. Una de las teorías es el incremento del cotransportador de K-Cl, el cual es activado por el pH ácido. En la EH, los hematíes, particularmente de pacientes no esplenectomizados, tienen un pH intracelular disminuido que es reflejo del pH bajo en el medio esplénico. El cotransportador de K-Cl también es activado por el daño oxidativo. Finalmente, la superactividad de bomba Na+-K+ ATPasa provocada por el incremento intracelular de sodio deshidrata la célula directamente, porque 3 iones Na+ son intercambiados por solo 2 iones K+. Esta pérdida de cationes monovalentes de sodio es acompañada por agua.1,2
]]>
Lípidos de la membrana
La principal anormalidad en la EH es la pérdida simétrica de cada tipo de lípido de membrana, como parte de la pérdida total de la superficie de membrana (el elemento distintivo de la fisiopatología de la EH). La proporción relativa del colesterol y de varios de los fosfolípidos son normales y estos últimos muestran la usual asimetría transmembrana, incluso en los casos más severos.
FUNCIÓN DEL BAZO
El bazo es muy importante en la fisiopatología de la EH. La disminución de la deformabilidad de los eritrocitos anormales y su destrucción esplénica, es la causa fundamental de la hemólisis. ]]>
Atrapamiento esplénico de los esferocitos no deformables
La anatomía única de la vasculatura esplénica lleva a un secuestro selectivo de esferocitos en el bazo. La sangre arterial entra en los cordones esplénicos, una red de vías constituidas por células reticulares y forradas por macrófagos. La mayoría de esta sangre pasa rápidamente a través de canales directos, los que reingresan en el sistema venoso luego de atravesar las fenestraciones. Una pequeña fracción de sangre percola en los cordones esplénicos más lentamente a través de la masa antes de reincorporarse a los sinusoides venosos. El tamaño de las fenestraciones en los sinusoides venosos es pequeño en comparación con el tamaño de los hematíes, por lo que para pasar a través de estos se requiere una deformabilidad significativa del eritrocito y su membrana.1
Debido a la disminución de la deformabilidad del esferocito, los hematíes son incapaces de atravesar las hendiduras entre el endotelio y las células adventicias que forman una pared, con lo que separan los cordones esplénicos de la pulpa roja. La reducción de la deformabilidad está relacionada principalmente con la disminución del área de superficie y secundaria al incremento de la viscosidad interna, que resulta en una moderada deshidratación celular. Adicionalmente, el medio esplénico es hostil para el eritrocito. Los niveles bajos de pH, glucosa y concentración de ATP, y las altas concentraciones locales de radicales libres tóxicos producidas por los fagocitos adyacentes, contribuyen al daño de membrana.2
]]>
Condicionamiento y destrucción de los esferocitos en el bazo
Los hematíes son "condicionados" en el bazo, incrementan su fragilidad osmótica y su condición esférica, con un ingreso neto de sodio y potasio más bajo que el obtenido por los hematíes en la circulación sistémica. El condicionamiento esplénico es una consecuencia de múltiples episodios de éstasis esplénico. El tiempo de permanencia estimado de los hematíes de la EH en los cordones es entre 10 y 100 minutos. Solo del 1 al 10 % de la sangre que entra en el bazo es detenida por los cordones congestionados, mientras que más del 90 % es rápidamente desviada a la circulación venosa.1,2
La fagocitosis por los macrófagos en el bazo es el paso final en el ciclo de la destrucción de los esferocitos. El estímulo para la fagocitosis por los macrófagos sigue siendo desconocido.
]]>
Herencia
Los genes responsables de la EH incluyen los de la ankirina, banda 3, a y b espectrinas y proteína 4.2. Alrededor de dos tercios de los pacientes con EH tienen un patrón de herencia autosómico dominante y es más frecuente la deficiencia de la ankirina.44 En los pacientes restantes donde el patrón dominante no puede ser demostrado, el patrón puede ser autosómico recesivo o resultar una mutación de novo.
Casos con herencia autosómica recesiva resultan de defectos en la a espectrina o la proteína 4.2. Se ha reportado un gran número de nuevas mutaciones en los genes de la EH.31,33,39,45,46 Se ha descrito un pequeño grupo de casos homocigóticos o dobles heterocigóticos para defectos en la banda 3 o las espectrinas, lo que lleva a la muerte fetal o anemia severa en el período neonatal.
En general, los individuos afectados con el mismo compromiso genético tienen similar grado de hemólisis.2,47
Cuando se identifica la EH en uno o más hermanos a los que los padres no le fueron identificadas anormalidades, o presentan gran variabilidad clínica en los miembros afectados de la misma familia, las causas pueden estar relacionadas con: herencia de un alelo modificado que influye en la expresión de las proteínas de membrana, penetrancia variable del defecto genético, una mutación de novo, una forma moderada de un patrón de herencia recesivo o un mosaicismo del defecto.48
NUEVOS POSTULADOS ]]>
Tradicionalmente, el compromiso de las proteínas de membrana y el efecto del bazo sobre los hematíes anómalos han sido los 2 grandes pilares manejados en la base de la fisiopatología de la EH. En la actualidad, con el desarrollo de las investigaciones, parecen estar involucrados nuevos procesos en la enfermedad.
Durante la enucleación del eritroblasto, las proteínas de la membrana plasmática y el citoesqueleto se reorganizan dinámicamente, mientras el núcleo se separa del reticulocito naciente. Un aspecto importante en este proceso es la división que sufren las proteínas de la membrana para que sea posible la salida del núcleo, por lo que la caracterización de las proteínas durante la enucleación es vital en la determinación del contenido de proteínas resultante en la membrana y citoesqueleto del reticulocito.49
Las proteínas pudieran no estar ensambladas correctamente en la membrana del eritroblasto y el orden durante la enucleación pudiera perturbarse o las proteínas sufrir una degradación intracelular o pérdida durante la maduración del reticulocito.49
Otros estudios confirman disturbios en el proceso de eritropoyesis relacionados con un componente inflamatorio, que pudiera, incluso, modular la severidad en la expresión clínica de la enfermedad. Lo anterior se basa en investigaciones donde se han demostrado valores significativamente elevados de eritropoyetina, receptor soluble de la transferrina, reticulocitos e índice de producción de reticulocitos, pero solo en el caso de la EH moderada se observaron niveles normales de hemoglobina. Esta correlación positiva entre la eritropoyetina, el receptor soluble de la transferrina, los reticulocitos y el índice de producción eritrocitaria observada en la EH moderada, no se encontró en pacientes con mayor severidad de la enfermedad, los que presentaron marcados niveles de neutrófilos, factor de necrosis tumoral a, interferón g, elastasa, lactoferrina y ferritina. Estos marcadores de inflamación pudieran frenar el efecto de la eritropoyesis en las formas más severas.50
A la luz de los conocimientos actuales, los fundamentos fisiopatológicos de la EH responden a un proceso multifactorial en el que se destacan como pilares básicos el compromiso molecular de las proteínas que conforman la membrana y el condicionamiento esplénico. Cada día se describen nuevas mutaciones moduladoras de la expresión clínica de la enfermedad, en consonancia con otros fenómenos eritropoyéticos, genéticos e inflamatorios. El conocimiento minucioso de cada uno de los elementos involucrados en la patogénesis de la EH garantizará un mejor diagnóstico y manejo de la enfermedad.
REFERENCIAS BIBLIOGRÁFICAS ]]>
1. Gallagher PG, Glader B. Hereditary spherocytosis, hereditary elliptocytosis, and other disorders associated with abnormalities of the erythrocyte membrane. En: Wintrobe's Clinical Hematology.12th ed. Philadelphia: Lippincott Williams & Wilkins; 2009. p. 912-30.
2. Kaushansky K, Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Prchal JT. The red blood cell membrane and its disorders: Hereditary spherocytosis, elliptocytosis, and related diseases. En: Kaushansky K, Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Prchal JT. William´s Hematology. 8th ed. New York: McGraw-Hill; 2010.
3. Holthuis JC, van Meer G, Huitema K. Lipid microdomains, lipid translocation and the organization of intracellular membrane transport. Mol Membr Biol. 2003 Jul-Sept;20(3):231-41.
]]>
4. Murphy SC, Samuel BU, Harrison T, Speicher KD, Speicher DW, Reid ME, et al. Erythrocyte detergent-resistant membrane proteins: their characterization and selective uptake during malarial infection. Blood. 2004 Mar;103(5):1920-8.
5. Mohandas N, Gallagher P. Red cell membrane: past, present, and future. Blood. 2008 Nov; 112(10):3939-48.
6. Diez-Silva M, Dao M, Han J, Lim CT, Suresh S. Shape and biomechanical characteristics of human red blood cells in health and disease. MRS Bull. 2010 May;35(5):382-8.
7. Pasini EM, Kirkegaard M, Mortensen P, Lutz HU, Thomas AW, Mann M. In-depth analysis of the membrane and cytosolic proteome of red blood cells. Blood. 2006 Aug;108(3):791-801.
8. Zhang Y, Zhang Y, Adachi J, Olsen JV, Shi R, de Souza G, et al. MAPU: Max-planck unified database of organellar, cellular, tissue and body fluid proteomes. Nucleic Acids Res. 2007 Jan; 35(suppl1):D771-D779. doi:10.1093/nar/gkl784.
9. Fairbanks G, Steck TL, Wallach DF. Electrophoretic analysis of the major polypeptides of the human erythrocyte membrane. Biochemistry. 1971 Jun;10(13):2606-17.
10. Gayani C, Spector J, Sullivan C, Kuypers FA, Labotka R, Gallagher P, et al. Imaging of the diffusion of single band 3 molecules on normal and mutant erythrocytes. Blood. 2009 June; 113(24):6237-45. ]]>
11. Beutler J, Mohandas N, Waugh RE. Integral protein linkage and the bilayer-skeletal separation energy in red blood cells. Biophys J. 2008 Aug;95(4):1826-36.
12. Ipsaroc JJ, Huang l, Mondrag A. Structures of the spectrin-ankyrin interaction binding domains. Blood. 2009 May;113(22):5385-93.
13. Stabach PR, Simonovic I, Ranieri MA, Aboodi MS, Steitz TA, Simonovic M, et al. The structure of the ankyrin-binding site of beta-spectrin reveals how tandem spectrin-repeats generate unique ligand-binding properties. Blood. 2009 May 28;113(22):5377-84.
]]>
14. Satchwell TJ, Shoemark DK, Sessions RB, Toye AM. Protein 4.2: A complex linker. Blood Cell Molecules Dis. 2009 May-Jun;42(3):201-10.
15. Bustos SP, Reithmeier RA. Protein 4.2 interaction with hereditary spherocytosis mutants of the cytoplasmic domain of human anion exchanger 1. Biochem J. 2010 Dec;433(2):313-22.
16. Ipsaro JJ, Mondrag A. Red Cells, Iron and erythropoiesis structural basis for spectrin recognition by ankyrin. Blood. 2010 May;115(20):4093-4101.
17. Deplaine G, Safeukui I, Jeddi F, Lacoste F, Brousse V, Perrot S, et al. The sensing of poorly deformable red blood cells by the human spleen can be mimicked in vitro. Blood. 2011 Feb;117(8):88-95.
18. Stadnick H, Onell R, Acker JP, Holovati JL. Eadie-Hofstee analysis of red blood cell deformability. Clin Hemorheol Microcirc. 2011;47(3):229-39.
19. Chakravarty S, Rizvi SI. Circadian modulation of sodium-potassium ATPase and sodium- proton exchanger in human erythrocytes: in vitro effect of melatonin. Cell Mol Biol. 2011 Feb;57(1):80-6.
20. Quarmyne MO, Risinger M, Linkugel A, Frazier A, Joiner C. Volume regulation and KCl cotransport in reticulocyte populations of sickle and normal red blood cells. Blood Cells Mol Dis. 2011 Aug;47(2):95-9. ]]>
21. Adragna NC, Fulvio MD, Lauf PK. Regulation of K-Cl cotransport: From function to genes. J Membr Biol. 2004 Oct 1;201(3):109-37.
22. Carbrey JM, Agre P. Discovery of the aquaporins and development of the field. Handb Exp Pharmacol. 2009;(190):3-28. PMID:19096770.
23. Blanc L, Liu J, Vidal M, Chasis JA, An X, Mohandas N. The water channel aquaporin-1 partitions into exosomes during reticulocyte maturation: implication for the regulation of cell volumen. Blood. 2009 Oct;114(18):3928-34.
]]>
24. Swietach P, Tiffert T, Mauritz JM, Seear R, Esposito A, Kaminski CF, et al. Hydrogen ion dynamics in human red blood cells. J Physiol. 2010 Dec;588(Pt 24):4995-5014.
25. Combet S, Zanotti JM, Bellissent-Funel MC. Temperature- and hydration-dependent internal dynamics of stripped human erythrocyte vesicles studied by incoherent neutron scattering. Biochim Biophys Acta. 2011 Feb;1810(2):202-10.
26. Debaugnies F, Cotton F, Boutique C, Gulbis B. Erythrocyte membrane protein analysis by sodium dodecyl sulphate-capillary gel electrophoresis in the diagnosis of hereditary spherocytosis. Clin Chem Lab Med. 2011 Mar;49(3):485-92.
27. Peker S, Akar N, Demiralp DO. Proteomic identification of erythrocyte membrane protein deficiency in hereditary spherocytosis. Mol Biol Rep. 2012 Mar;39(3):3161-7. DOI: 10.1007/s11033-011-1082-x.
28. Iolascon A, Avvisati RA .Genotype/phenotype correlation in hereditary spherocytosis. Haematologica. 2008;93(9):1283-7.
29. Huq S, Pietroni M, Rahman H, Tariqul M. Hereditary spherocytosis. J Health Popul Nutr. 2010 Feb;28(1):107-9.
30. Perrotta S, Della Ragione F, Rossi F, Avvisati RA, Di Pinto D, De Mieri G, et al. B-spectrinBari: a truncated b-chain responsible for dominant hereditary spherocytosis. Haematologica. 2009;94(12)1753-7. ]]>
31. Maciag M, Ochocka D, Adamowicz-Salach A, BurzyÅska B. Novel beta-spectrin mutations in hereditary spherocytosis associated with decreased levels of mRNA. Br J Haematol. 2009 Aug;146(3):326-32.
32. Gundel F, Eber S, Heep A. A new ankyrin mutation (ANK1 EXON E9X) causing severe hereditary spherocytosis in the neonatal period. Ann Hematol. 2010;90(2):231-2.
33. Hughes MR, Anderson N, Maltby S, Wong J, Berberovic Z, Connie S, et al. A novel ENU-generated truncation mutation lacking the spectrin-binding and C-terminal regulatory domains of Ank1 models severe hemolytic hereditary spherocytosis. Experimental Hematology. 2011 March;39(3) 305-20.
]]>
34. Gallagher PG, Steiner LA, Liem RL, Owen A, Cline AP, Seidel N, et al. Mutation of a barrier insulator in the human ankyrin-1 gene is associated with hereditary spherocytosis. J Clin Invest. 2010 Dec;120(12):4453-65.
35. An X, Mohandas N. Disorders of red cell membrane. Br J Haematol. 2008 May;141(3):367-75.
36. Ribeiro ML, Alloisio N, Almeida H, Gomes C, Texier P, Lemos C, et al. Severe hereditary spherocytosis and distal renal tubular acidosis associated with the total absence of band 3. Blood. 2000 Aug 15;96(4):1602-4.
37. Wrong O, Bruce LJ, Unwin RJ, Toye AM, Tanner MJ. Band 3 mutations, distal renal tubular acidosis, and Southeast Asian ovalocytosis. Kidney Int. 2002 Jul;62(1):10-9.
38. Sánchez-López JP, Camacho-Torres AL, Ibarra B, Tintos JA, Perea FJ. Analysis of the SLC4A1 gene in three Mexican patients with hereditary spherocytosis: Report of a novel mutation. Genet Mol Biol. 2010 Jan-Mar;33(1):9-11.
39. Chu C, Woods N, Sawasdee N, Guizouarn H, Borgese B, Yenchitsomanus P, et al. Band 3 Edmonton I, a novel mutant of the anion exchanger 1 causing spherocytosis and distal renal tubular acidosis. Biochem J. 2010 Feb 24;426(3):379-88.
40. Bouhassira EE, Schwartz RS, Yawata Y, Ata K, Kanzaki A, Qiu JJ, et al. An alanine-to-threonine substitution in protein 4.2 cDNA is associated with a Japanese form of hereditary hemolytic anemia (protein 4.2NIPPON). Blood. 1992 Apr;79(7):1846-54. ]]>
41. Yawata Y, Kanzaki A, Yawata A, Doerfler W, Ozcan R, Eber SW. Characteristic features of the genotype and phenotype of hereditary spherocytosis in the Japanese population. Int J Hematol. 2000 Feb;71(2):118-35.
42. Satchwell TJ, Shoemark DK, Sessions RB, Toye AM. Protein 4.2: a complex linker. Blood Cells Mol Dis. 2009 May-June;42(3):201-10.
43. Akker E, Satchwell TJ, Pellegrin S, Flatt JF, Maigre M, Daniels G, et al. Investigating the key membrane protein changes during in vitro erythropoiesis of protein 4.2 cells (mutations Chartres 1 and 2). Haematologica. 2010 Aug;95(8):1278-86.
]]>
44. Tracy ET, Rice HE. Partial splenectomy for hereditary spherocytosis. Pediatr Clin N Am. 2008;55:503-19.
45. Miraglia del Giudice E, Francese M, Nobili B, Morlé L, Cutillo S, Delaunay J, et al. High frequency of de novo mutations in ankyrin gene (ANK1) in children with hereditary spherocytosis. J Pediatr. 1998 Jan;132(1):117-20.
46. Miraglia del Giudice E, Lombardi C, Francese M, Nobili B, Conte ML, Amendola G, et al. Frequent de novo monoallelic expression of beta-spectrin gene (SPTB) in children with hereditary spherocytosis and isolated spectrin deficiency. Br J Haematol. 1998 May;101(2):251-4.
47. Rocha S, Costa E, Rocha-Pereira P, Ferreira F, Cleto E, Barbot J, et al. Erythrocyte membrane protein destabilization versus clinical outcome in 160 Portuguese Hereditary Spherocytosis patients. Br J Haematol. 2010 Jun;149(5):785-94. ]]>
48. Eber SW, Armbrust R, Schroter W. Variable clinical severity of hereditary spherocytosis: Relation to erythrocytic spectrin concentration, osmotic fragility, and autohemolysis. J Pediatr. 1990 Sep;117(3):409-16.
49. Salomao M, Chen K, Villalobos J, Mohandas N, An X, Chasis JA. Hereditary spherocytosis and hereditary elliptocytosis: aberrant protein sorting during erythroblast enucleation. Blood. 2010 July;116(2):267-69.
50. Rocha S, Costa E, Rocha-Pereira P, Ferreira F, Cleto E, Barbot J, et al. Erythropoiesis versus inflammation in hereditary spherocytosis clinical outcome. Clin Biochem. 2011 Sep;44(13):1137-43.
]]>
Recibido: 15 de agosto del 2012.
Aprobado: 15 de septiembre del 2012.
]]>
Dra. Heidys Garrote-Santana. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800. La Habana, Cuba. Tel (537) 643 8695, 8268, Fax (537) 644 2334. Correo electrónico: rchematologia@infomed.sld.cu
Website: http://www.sld.cu/sitios/ihi ]]>
{kind=link}