REVIEW
Excitotoxicity and neuronal death in epilepsy
Excitotoxicidad y muerte neuronal en la epilepsia
]]>
Lourdes Lorigados1, Sandra Orozco2, Lilia Morales3, Bárbara Estupiñán3, Iván García4, Luisa Rocha5
1 Departamento de Inmunoquímica, Centro Internacional de Restauración Neurológica, Cirén. Ave. 25, No. 15805 e/ 158 y 160 Playa, CP 11300, La Habana, Cuba.
2 Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social, IMSS, México.
3 Departamento de Neurofisiología, Centro Internacional de Restauración Neurológica, Cirén. La Habana, Cuba.
4 Servicio de Neurocirugía, Centro Internacional de Restauración Neurológica, Cirén. La Habana, Cuba.
5 Laboratorio Farmacobiología, Centro de Investigación y de Estudios Avanzados, Cinvestav, Sede Sur, DF, México.
Epilepsy is a recurrent, often progressive neurological disorder with a chronic evolution, affecting 1 to 2 % of the world population. Research with experimental models and imaging analysis of diseased patients have been used to show that recurrent episodes produce oxidative stress, most of which is related to neuronal excitability phenomena. It is known that the excessive stimulation of glutamate receptors results in neurotoxicity; a process that, under the denomination of excitotoxicity, is thought to constitute the principal cellular death mechanism behind different disorders of the central nervous system, including epilepsy. Paradoxically, although the signaling pathways, molecular mechanisms and sites of action of excitotoxicity have received considerable attention since the 1970s, little is known about their relevance to CNS disorders. Further detail is necessary about the fundamental role of neuronal death and the mechanisms, particularly those relevant to neurological pathogenesis, that are engaged whenever glutamate receptors are excessively stimulated, as the results would aid considerably the development of timely clinical interventions delaying the evolution of these disorders. We review clinical and experimental data on the relevant alterations of the glutamatergic system, cell death pathways, and the activation of caspases and members of the Bcl-2 gene family involved in the process as modulators of cell death during epilepsy. The findings support the hypothesis that excitotoxic processes as well as both apoptotic and necrotic neuronal cell death phenomena converge in drug-resistant epilepsy.
Keywords: excitotoxicity, apoptosis, necrosis, epilepsy.
RESUMEN
La epilepsia es una afección neurológica de evolución crónica, recurrente, casi siempre progresiva, que afecta del 1 al 2 % de la población mundial. Modelos experimentales y estudios de imágenes neurológicas de pacientes con este padecimiento muestran que las crisis recurrentes provocan estrés oxidativo, relacionado fundamentalmente con la excitabilidad neuronal. La estimulación excesiva de los receptores de glutamato induce neurotoxicidad, un proceso que se ha definido como excitotoxicidad. Se considera que este puede ser el principal mecanismo de muerte celular en numerosas afecciones del sistema nervioso central, incluida la epilepsia. Desde los años 70 se han estudiado con profundidad las vías de señalización, los mecanismos moleculares y los sitios de acción relacionados con la excitotoxicidad; aunque de forma muy limitada en las enfermedades del sistema nervioso central. En particular, deberán evaluarse con especial cuidado la función crucial de la muerte neuronal y los mecanismos que se potencian con la sobreactivación de los receptores de glutamato, principalmente los relativos a las enfermedades neurológicas, con el fin de intervenir de manera oportuna para retardar el desarrollo de estas afecciones neurológicas. Se repasan las evidencias clínicas y experimentales sobre las alteraciones del sistema glutamatérgico, las vías de muerte celular, la activación de las caspasas y de la familia de genes Bcl-2 involucrados, como moduladores de la muerte celular en la epilepsia. Tales hallazgos sustentan que en la epilepsia farmacorresistente convergen procesos excitotóxicos y de muerte neuronal apoptótica y necrótica.
Palabras clave: excitotoxicidad, apoptosis, necrosis, epilepsia.
]]>
INTRODUCTION
Glutamate receptor-mediated excitotoxicity not only plays an important role in neural development, differentiation and synaptic plasticity [1, 2], but is regarded as the principal mechanism for cell death in a number of disorders of the central nervous system (CNS), including brain trauma, neurodegenerative disorders and epilepsy [3-6].
Glutamate is the ultimate excitatory neurotransmitter of mammalian CNS. Accurate control of glutamatergic neurotransmission is of paramount importance, due to its involvement in both excitotoxic cell death and neural signaling [2]. Early descriptions of excitotoxicity-mediated cell death mentioned increases in cell volume, vacuolization of the cytoplasm and loss of membrane integrity; all of which are consistent with a necrotic mechanism for this event [7-10]. Later evidence, however, has demonstrated that this process can also be associated with apoptotic hallmarks such as the degradation of DNA at internucleosomal sites and the activation of caspases [11-13]. In addition, recent publications have pointed at autophagy, induced as the result of sustained cellular stress, as the mechanism behind excitotoxicity-induced non-apoptotic cell death [2]. Increased glutamate receptor activity levels would, therefore, induce the expression of pro-apoptotic proteins such as p53, leading to cell damage and death mediated by apoptosis or autophagy [14-16]. The latter would be induced as a response to acute excitotoxic damage [17, 18].
Despite the large body of knowledge accrued on the signaling pathways and the sequence of events taking place during excitotoxicity, little is known about its role in the CNS or the molecular mechanisms underlying its effects. It is clear, however, that far from being a uniform process, excitotoxic cell death in the brain actually represents a continuum going from necrosis to apoptosis and autophagy. This review discusses the cellular and molecular mechanisms of excitotoxicity and its effect on the process of neuronal death taking place during epilepsy.
CONCEPT OF EXCITOTOXICITY
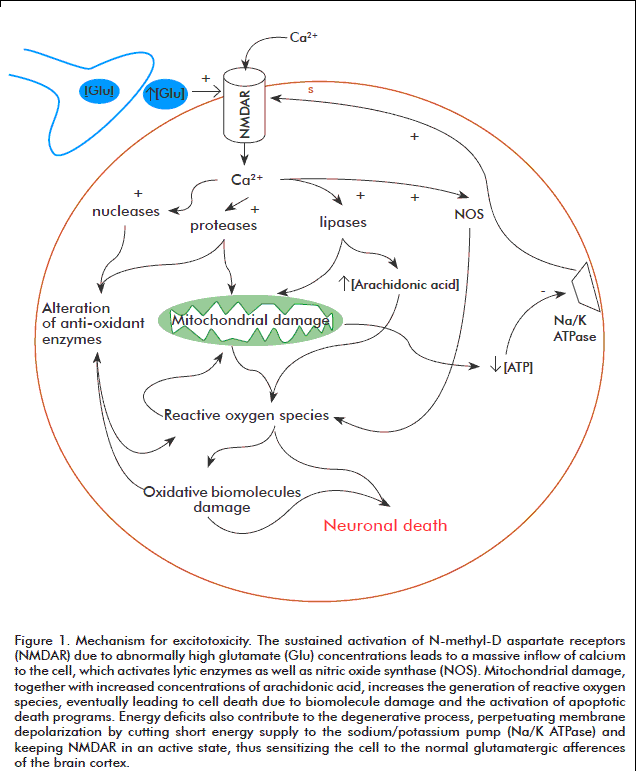
A number of different experimental and clinical findings on the potential toxicity of excitatory aminoacids have provided the foundation for excitotoxic theory, which postulates the existence of a direct link between neuronal degeneration and glutamate receptor hypersensitivity or excessive levels of endogenous glutamate [19]. Excitotoxicity is therefore a mechanism promoting cell death through the hyper activation of glutamatergic receptors or its analogues. This hyper activation leads to excess calcium (Ca2+) inflow to the cell, where this ion is sequestered inside mitochondria, leading to metabolic dysfunction, the generation of free radicals, the activation of proteases, phospholipases, endonucleases, nitric oxide synthase, and the inhibition of protein synthesis [20].
For calcium homeostasis to be lost, regulatory mechanisms for this ion, including the calcium pump, the sodium/calcium (Na+/Ca2+) exchanger and calcium buffering proteins, must first be overflowed. Once these systems saturate, excess calcium accumulates inside the mitochondrial matrix. This accumulation depolarizes the mitochondrial membrane by two different mechanisms: first, the increased concentration of positive ions in the mitochondrial matrix decreases the chemo-osmotic potential across the membrane (leading in turn to reduced rates of adenosine triphosphate (ATP) synthesis), and second, the activation of mitochondrial transition pores (a mechanism normally used to shunt calcium back to the cytosol), which can lead to irreversible membrane depolarization if calcium unbalance is prolonged [21, 22]. High calcium concentrations in the mitochondrial matrix also promote the generation of free radicals, which promote the peroxidation of membrane lipids, the synthesis of nitric oxide and the activation of enzymes involved in the catabolism of proteins, phospholipids and nucleic acids. In addition, nitric oxide can act as a retrograde messenger, further contributing to the excitotoxic effect of glutamate by enhancing its release from pre-synaptic terminals [23] (Figure 1).
]]> Additional contributions to cellular damage are provided by the activation of nitric oxide synthase, whose reaction products react with superoxide anions to yield peroxynitrite, and the activation of poly-adenosine diphosphate ribose-polymerase (PARP), triggered by free radical-mediated DNA damage [24, 25].
EXCITOTOXICITY AND EPILEPSY
There is evidence supporting the hypothesis that the neurodegenerative changes associated with human epilepsy arise from persistent discharges in the glutamate pathway. The mechanism is relatively simple: excess glutamate release leads to repeated depolarization-repolarization cycles in glutamate terminals, until glutamate reaches toxic concentrations and, finally, the excitotoxic degeneration of post-synaptic neurons takes place [26, 27].
Micro-dialysis studies in humans and animal models have demonstrated an association between prolonged convulsive activity and the duration of the epileptic episode due to significant increases in glutamate levels [28]. It is well known that neuronal over excitation by glutamate can trigger epileptic seizures, and that the effect of directly applying glutamate to the amygdala is similar to that of propagated activation [29]. Using agonists of the α-amino-3-hydroxy-5-methyl-4-isoxazolpropionic acid receptor has been shown to delay the development of propagated activation at the amygdala in mice [30].
The activation of N-methyl-D-aspartate receptors is a mediator of cell death during the epileptic state [31], and the use of MK-801, an antagonist for this receptor, prevents the occurrence of spontaneous seizures in animal models [32]. Kainate receptors, specifically the GluR6 subunit, are known to participate in epileptogenesis as inducers [33, 34].
In general, excitotoxic damage to the neurons of epileptic patients is mediated by excessive calcium inflow during seizures [35]. The resulting high levels of calcium trigger a sequence of events that includes the activation of nitric oxide synthase, thereby interfering with oxidative metabolism and generating free radicals that ultimately damage the neuronal membrane. Pro-caspases are activated likewise, and neuronal death eventually takes place by necrosis, apoptosis or autophagy.
]]> EXCITOTOXICITY IN EXPERIMENTAL MODELS OF EPILEPSY
The limitations for studying epilepsy in humans have led to the development of experimental models reproducing this condition. It should be noticed, however, that existing models fail to accurately reproduce the behavioral manifestations of this disease, especially in the case of motor alterations.
Animal epilepsy models are classified as either acute or chronic. The former are implemented through the delivery of convulsant drugs or the application of electrical stimulation; the latter, while harder to implement, provide a closer approximation to the physiopathology of this disorder in humans, although both are capable of producing partial and generalized seizures. In addition, true epilepsy models reproduce the recurrence of ictal manifestations characterizing this disease in humans. The ultimate challenge when using experimental models to study epilepsy is to determine which, of the many alterations stemming from a specific brain injury, is causally linked to the subsequent development of epilepsy.
Oxidative stress plays an important role in cellular damage and death induced by recurrent seizures. The free radicals generated during oxidative stress have long been acknowledged as part and parcel of excitotoxicity, due in part to the fact that prolonged, sustained seizures generate macromolecular damage in the cells that is related, above all, to neuronal excitability.
Animal models have been used to study two specific types of epileptic events: prolonged (20 to 30 min) febrile crises and long epileptic seizures (5 to 6 h), the latter induced through the systemic delivery of cholinergic agonists (pilocarpine) or by unilateral injection of glutamatergic agonists into the hippocampus of experimental rats (kainic acid, a glutamate analog). Using models of febrile crises, neonatal hypoxia and spasms, it has been possible to demonstrate that developing neurons are less vulnerable to cellular damage and survive much better than fully grown neurons. For instance, the hippocampal neurons of animals with immature brains placed under an anoxic environment continue to react to synaptic stimuli for a longer time and require more prolonged exposures to completely and irreversibly destroy their neural circuits [36]. Immature brains also appear to be more resistant to the toxic effects of glutamate than mature ones [37]. Mark et al. [38] demonstrated that the amount of calcium entering a pyramidal neuron is directly related to the animal’s age: during the first three days of life, glutamate increases calcium concentration only by marginal amounts; however, in days 21 to 25 there is a marked increase in intracellular calcium concentration and soma volume, while dendrites retract. This relatively higher resistance stems from a lower density of active synapses, lower energy requirements and, in general, the relative immaturity of the biochemical cascades leading to cell death, explaining why young individuals are less vulnerable than adults to the cell loss taking place after prolonged epileptic seizures [39-41].
The most popular models of excitotoxicity employing adult animals are those based on the use of kainic acid and pilocarpine. These are models of epilepsy of the temporal lobe, induced by the unilateral or systemic injection of these compounds at convulsant doses, causing excitotoxic damage at the pyramidal neurons of hippocampus and the hilar region. Damage depends on dosage, species and line of the animal, but the result in all cases is neuronal death at vulnerable regions, the proliferation of astrocytes and increased glial fibers. For these reasons, the models based on systemically administering kainic acid or pilocarpine are widely used for studying generalized tonic-clonic convulsions or the epileptic state, whose neuroanatomical substrate is temporal mesial sclerosis [42-46].
One of the first changes taking place after the injection of kainic acid is the induction of messenger RNA (mRNA) coding for heat shock proteins (HSPs) of varying molecular weights (HSP27, HSP70 and HSP72), whose expression levels increase consequently. HSP72, particularly, is constitutively expressed in the mammalian brain, and exhibits increased concentrations among sensitive neuronal populations of the hippocampus [47]. The expression of these chaperones seems to prevent the misfolding of newly synthesized proteins in kainic acid-vulnerable populations. During the two weeks following administration of the convulsant, these proteins are transported to the most distal zones along dendrites and axons. Both HSP70 and HSP72 have been shown to play a protective role in this process, although they are unable to rescue damaged cells from excitotoxic death. While the overexpression of HSP27 and HSP70 in vivo protects from excitotoxic damage [47, 48], excessively high levels of HSP72 can be noxious to the cell [49-51].
Three to five hours after the injection of kainic acid, mRNA coding for cFos and cJun are also induced, increasing the concentrations of their respective proteins at vulnerable regions of the hippocampus and dentate gyrus [52]. cFos immunoreactivity at the dentate gyrus disappears after six hours, but remains high at the hippocampus, suggesting an association of cell death with high cFos levels. However, a prolonged increase in cFos is poorly predictive, and is not a necessary condition for neuronal excitotoxic damage to take place [53, 54]. cJun levels have also been found to be high at the hippocampus and the dentate circumvolution for 24 hours after an epileptic seizure. The meaning of this increase is unclear, as cJun is considered both a marker of delayed cell death after epileptic seizures, and a potential marker for neuronal survival upon excitotoxic damage [52].
Regarding signaling mechanisms across the cell membrane, it has been observed that tissue plasminogen activator (tPA), an extracellular serine protease, appears to be necessary for cell death to take place, as knock-out tPA or plasminogen mice are relatively resistant to excitotoxicity. This effect seems to be mediated by the interaction of tPA with laminin, an extracellular matrix protein [55]. In addition, it has been shown that increases in the expression of the specific Fas receptor ligand (FasL) at hippocampus and granule cells of the dentate gyrus, three hours after the injection of kainic acid, are related with signaling across the cell membrane [56]. FasL expression at the granule cells of the dentate gyrus decreases six hours after the administration of kainic acid, although FasL immunoreactivity remains high at the hippocampus. The relevance of the latter finding is highlighted by the fact that binding of FasL to Fas activates its death domain, to which the Fas-associated death domain (FADD) then binds, activating in turn caspase 8, which acts upon effector caspases leading to cell death by apoptosis. The availability of Fas transgenic mice has been invaluable for examining the role of the Fas/FasL system in excitotoxic cell death signaling [56].
The role of members of the bcl-2 gene family in this process remains to be elucidated. A preliminary study observed reductions in the concentration of the Bcl-2 protein and increases in Bax mRNA in mouse hippocampus after the systemic administration of kainic acid [57]. Finer studies employing Northern blotting have detected that Bax mRNA (but not those coding for Bcl-2 and Bcl-x) is induced from hour 6 to 24 at the hippocampus of rats receiving kainic acid. The hippocampal concentration of Bcl-2, Bcl-x and Bax, measured by Western blotting and immunohistochemistry, is similar in cells that eventually die and in survivors [58]. Possibly, the effects exerted by members of the Bcl-2 family do not depend on global changes at the protein level, but rather on changes of their subcellular location. Apoptotic cell signaling through the mitochondrial pathway is triggered by Bax binding to the mitochondrial membrane and the release of cytochrome c to the cytosol, followed by binding to the apoptotic protease activation factor Apaf1 in the presence of ATP as well as the activation of caspase 9, which in turn activates different effector caspases. The mechanism whereby cytochrome c is released from the mitochondria into the cytosol remains unclear, but seems to require an interaction between Bcl-2, Bcl-x, Bax, and voltage-dependent ion channels controlling the release of cytochrome c. It has been argued that the Bax/Bcl-2 ratio plays an essential role in determining whether the cell enters apoptosis [59].
]]> Excitotoxicity resulting from the intraperitoneally delivery of kainic acid also induces the caspase 3 mRNA and increases procaspase 3 levels in some neurons at vulnerable regions of the hippocampus [60, 61]. Some neurons express the active (cleaved-off) 17 kDa fragment of caspase 3 [62], demonstrating the involvement of caspases in at least some hippocampal neurons and, therefore, the existence of an apoptotic component for cell death in hippocampal subpopulations. It should be noted, however, that Western blotting experiments have demonstrated the presence of PARP bands of 89 kDa and lower molecular weights, evidencing that fragmentation of the latter also involves proteases other than caspases and suggesting indiscriminate death by necrosis [61].One result of excitotoxic damage affecting preferentially hilar region cells [63] is that mossy fibers from the granule cells of the dentate gyrus become disconnected from their targets. This differentiation results in the sprouting of mossy axonal branches along the supragranular region and the molecular layer of the dentate gyrus [64, 65]. However, fiber sprouting after seizures is less evident in young animals [10, 66]. Fiber sprouting is associated with the expression of GAP-43 at the supragranular layer during the first week, and at the entire molecular layer after a month [67]. An increase of the 25 kDa synaptosome-associated protein (SNAP25) has been detected at neurons and the molecular layer of the dentate gyrus during the days following kainic acid-induced excitotoxic injuries [68, 69]. The participation of specific trophic signals in the development of these aberrant connections seems likely, but their function remains contentious. Brain-derived neurotrophic factor, as well as the TrkB receptor in dentate gyrus neurons, possibly modulate the trophism of these cells during the formation of plastic ramifications to re-innervate the zones destroyed by kainic acid. It has also been suggested that brain-derived neurotrophic factor plays a protective role by attenuating oxidative stress [70, 71].
NEURONAL DEATH AND EPILEPSY
Apoptosis is a characteristic form of programmed cell death that is under control of a common genetic program across differing cell types. It is usually triggered at the level of individual cells, rather than at the level of entire tissues. Among the first morphological changes cells exhibit when the apoptotic process starts are the condensation of their cytoplasm and cell shrinkage, together with changes in nuclear structure. Chromatin is condensed and forms dense, compact patches against the nuclear envelope, after which the latter forms invaginations and the nucleus is fragmented into membranous structures containing variable amounts of chromatin. In a similar manner, the cell membrane starts producing blebs and ends up fragmented in clusters of variable-sized vesicles containing intact organelles that are not fused with lysosomes. These vesicles, denominated apoptotic bodies, are quickly phagocytosed by neighboring cells. One of the most physiologically relevant consequences of neuron death by apoptosis is, therefore, that no intracellular material is released to the interstitial milieu [72].
Seizure-induced neuronal death does not negate the molecular complexity of neural death due to neurodegeneration. Whether neuronal death is apoptotic or necrotic remains controversial. Based on the classical definition and the morphological features typical of necrosis, this constitutes the most frequent mechanism of death for brain neurons after a seizure [7-10, 13, 73], and many authors argue that neuronal death by necrosis is the dominant post-seizure cell death mechanism [73, 74]. However, the involvement of apoptosis cannot be readily dismissed, as biochemical studies have established that members of the Bcl-2 family and caspases are involved in post-seizure cell death. In addition, DNA fragments whose sizes are multiples of 180-200 base pairs, early endonuclease activation and DNA fragmentation (originally described as apoptotic hallmarks) have all been found in cells programmed to die, and p53 has been shown to accumulate at the nucleus of kainic acid-vulnerable neurons, concurrently with increases in the concentration of cell death receptors and their ligands. Apoptotic mechanisms are, therefore, involved in the process of cell death [11-13, 75-78].
Alterations in the Bcl-2 family of proteins and the proteolytic cleavage of pro-caspases 1 and 3 have been described, in addition to the detection of several markers of apoptotic cell death in different experimental models of epilepsy: caspases are activated by seizures, as are neuronal death receptors and Bcl-2 family members [62, 76, 78-83]. Increases in serum Bcl-2 for patients with temporal lobe epilepsy have been found to correlate with the duration of the disease, seizure frequency and disorder severity [84].
Gene p53 was the first apoptotic regulator found to be damaged by seizures [85]. It has been shown that levels of both its mRNA and the protein itself increase, based functionally on 1) the binding of p53 to DNA takes place after the seizures [86], and 2) the expression of Bax increases with the seizures [57, 87]. A known inhibitor of p53 synthesis has been shown to protect against kainic acid excitotoxicity [88]. p53-defficient mouse neurons are resistant to seizures and apoptosis induced by excitotoxins [89]. However, the consequences of p53 alterations on seizure-induced neuronal death are not completely clear, due to the multiple roles of p53. In general, the data support that caspases, Bcl-2 and p53 all get involved in some form after seizures.
We have discussed the classical division into apoptosis or necrosis; two processes that can take place independently, sequentially and even simultaneously [79, 90], depending on stimulus type and intensity. A model is suggested providing continuity between the classical apoptotic cascade mediated by caspases and cellular necrosis or lysis [91]. Intermediate scenarios would be 1) programmed cell death, similar to apoptosis, 2) caspase-independent cell death and 3) programmed cell death, similar to necrosis. This criterion is important, especially for analyzing cell death during neurological processes [92].
]]> A study performed by our group on drug-resistant temporal lobe epilepsy patients demonstrated the involvement of both neuronal death processes (necrosis and apoptosis), as evidenced by increased immunoreactivity to Annexin V and by the results of a Tunnel assay in neocortical tissue (Figure 2). This indicated that cell death in this brain area might be apoptotic, without dismissing the possibility of necrotic cell death, as the Tunnel+ marker has been shown to associate with both processes [93]. In addition, we demonstrated the presence of redox unbalances in these patients [94], a phenomenon that would lead to cell death due to mitochondrial dysfunction caused by mitochondrial membrane depolarization. During later studies by electron microscopy, we were able to observe both necrotic and apoptotic cells in these tissues (Figure 3). These evidences may help in the development of neuroprotective strategies against the cell death processes triggered by epilepsy.
CONCLUSIONS
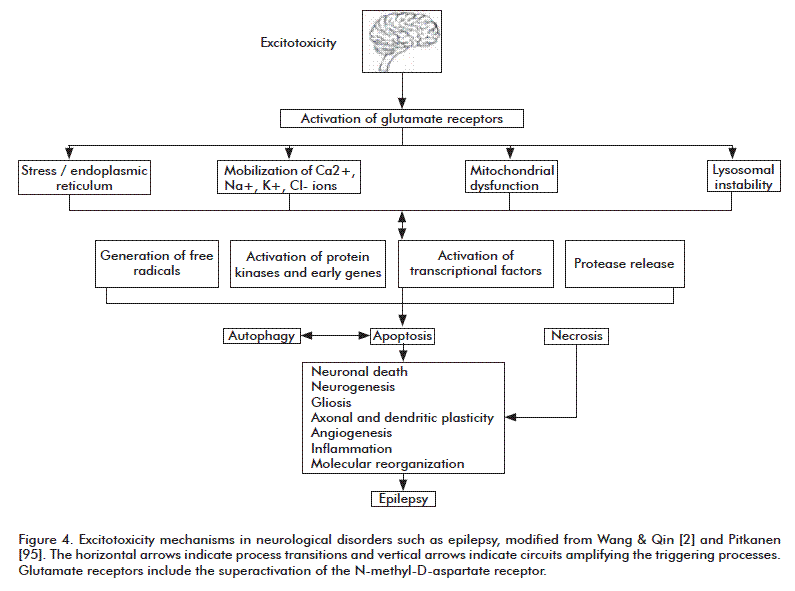
Contradictory findings regarding epilepsy abound in the experimental literature, due among other causes to the many models currently in use and difficulties inherent to trying to reproduce all the characteristics of this disease in different species. Studies in humans have targeted different locations of epileptogenic foci, times of evolution of the disorder, type and age at first seizure and other parameters. Further research is undoubtedly required to completely characterize the action of these cell death mechanisms during seizures, in order to establish an interaction procedure that can attenuate epileptic damage (Figure 4) summarizes the proposed mechanisms for excitotoxicity during neurological disorders.
Although the signaling pathways and sequence of events taking place during excitotoxicity have been extensively studied since the 1970s, knowledge about excitotoxicity in the CNS, its molecular mechanisms and action sites is still lacking. Careful evaluations of the essential roles of both neuronal death and the mechanisms recruited during overexcitation of glutamate receptors in neurological disorders are necessary to devise timely clinical interventions that can delay the development of diseases such as epilepsy. The most recent findings have demonstrated the convergence of excitotoxic processes together with apoptotic and necrotic neuronal death in drug-resistant epilepsy.
ACKNOWLEDGEMENTS
We would like to acknowledge the contributions of licentiates Leticia Neri Bazán and Héctor Vázquez Espinosa, as well as the support of the Unit for Medical Research on Neurological Disorders of the Specialty Hospital, 20th Century National Medical Center, belonging to the Mexican Welfare Institute (IMSS). This research was funded by Conacyt (project 98386).
]]>
REFERENCES
1. Yang JL, Sykora P, Wilson DM, 3rd, Mattson MP, Bohr VA. The excitatory neurotransmitter glutamate stimulates DNA repair to increase neuronal resiliency. Mech Ageing Dev. 2011;132(8-9):405-11.
2. Wang Y, Qin ZH. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis. 2010; 15(11):1382-402.
3. Severino PC, Muller Gdo A, Vandresen-Filho S, Tasca CI. Cell signaling in NMDA preconditioning and neuroprotection in convulsions induced by quinolinic acid. Life Sci. 2011;89(15-16):570-6.
4. Araujo IM, Carreira BP, Carvalho CM, Carvalho AP. Calpains and delayed calcium deregulation in ex-citotoxicity. Neurochem Res. 2010; 35(12):1966-9.
]]>5. Wang Y, Denisova JV, Kang KS, Fontes JD, Zhu BT, Belousov AB. Neuronal gap junctions are required for NMDA receptor-mediated excitotoxicity: implications in ischemic stroke. J Neurophysiol. 2010;104(6):3551-6.
6. Farooqui AA, Ong WY, Horrocks LA. Glutamate receptors and their association with other neurochemical parameters in excitotoxicity. In: Farooqui AA, Ong WY, Horrocks LA, editors. Neurochemical aspects of excitotoxicity. New York: Springer; 2008. p. 105-36.
7. Fujikawa DG, Shinmei SS, Cai B. Kainic acid-induced seizures produce necrotic, not apoptotic, neurons with internucleosomal DNA cleavage: implications for programmed cell death mechanisms. Neuroscience. 2000;98(1):41-53.
8. Fujikawa DG, Shinmei SS, Cai B. Seizure-induced neuronal necrosis: implications for programmed cell death mechanisms. Epilepsia. 2000; 41 Suppl 6:S9-13.
9. Ebert U, Brandt C, Loscher W. Delayed sclerosis, neuroprotection, and limbic epileptogenesis after status epilepticus in the rat. Epilepsia. 2002;43 Suppl 5:86-95.
]]>10. Kubova H, Druga R, Lukasiuk K, Suchomelova L, Haugvicova R, Jirmanova I, et al. Status epilepticus causes necrotic damage in the mediodorsal nucleus of the thalamus in immature rats. J Neurosci. 2001; 21(10):3593-9.
11. Bengzon J, Mohapel P, Ekdahl CT, Lindvall O. Neuronal apoptosis after brief and prolonged seizures. Prog Brain Res. 2002;135:111-9.
12. Henshall DC, Araki T, Schindler CK, Lan JQ, Tiekoter KL, Taki W, et al. Activation of Bcl-2-associated death protein and counter-response of Akt within cell populations during seizure-induced neuronal death. J Neurosci. 2002;22(19):8458-65.
13. Liou AK, Clark RS, Henshall DC, Yin XM, Chen J. To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: a review on the stress-activated signaling pathways and apoptotic pathways. Prog Neurobiol. 2003;69(2):103-42.
14. Dong XX, Wang Y, Qin ZH. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin. 2009;30(4):379-87.
]]>15. Qin ZH, Tao LY, Chen X. Dual roles of NF-kappaB in cell survival and implications of NF-kappaB inhibitors in neuroprotective therapy. Acta Pharmacol Sin 2007;28(12):1859-72.
16. Zhang XD, Wang Y, Zhang X, Han R, Wu JC, Liang ZQ, et al. p53 mediates mitochondria dysfunction-triggered autophagy activation and cell death in rat striatum. Autophagy. 2009;5(3):339-50.
17. Shacka JJ, Lu J, Xie ZL, Uchiyama Y, Roth KA, Zhang J. Kainic acid induces early and transient autophagic stress in mouse hippocampus. Neurosci Lett. 2007;414(1):57-60.
18. Wang Y, Dong XX, Cao Y, Liang ZQ, Han R, Wu JC, et al. p53 induction contributes to excitotoxic neuronal death in rat striatum through apoptotic and autophagic mechanisms. Eur J Neurosci. 2009; 30(12):2258-70.
19. Olney JW. Excitatory transmitters and epilepsy-related brain damage. Int Rev Neurobiol. 1985;27:337-62.
]]>20. Haglid KG, Wang S, Qiner Y, Hamberger A. Excitotoxicity. Experimental correlates to human epilepsy. Mol Neurobiol. 1994;9(1-3):259-63.
21. Nicholls DG, Ward MW. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci. 2000;23(4):166-74.
22. Murchison D, Griffith WH. Mitochondria buffer non-toxic calcium loads and release calcium through the mitochondrial permeability transition pore and sodium/calcium exchanger in rat basal forebrain neurons. Brain Res. 2000;854(1-2):139-51.
23. Almeida A, Heales SJ, Bolanos JP, Medina JM. Glutamate neurotoxicity is associated with nitric oxide-mediated mitochondrial dysfunction and glutathione depletion. Brain Res. 1998;790(1-2):209-16.
24. Olney JW. New insights and new issues in developmental neurotoxicology. Neurotoxicology. 2002;23(6):659-68.
]]>25. Struzynska L. A glutamatergic component of lead toxicity in adult brain: the role of astrocytic glutamate transporters. Neurochem Int. 2009;55(1-3):151-6.
26. Pereno GL. Fisiopatología de la epilepsia del lóbulo temporal: revisión del proceso de muerte neuronal a la neuroplasticidad. Rev Argentina Cienc Comportamiento. 2010;2(1):46-57.
27. Eid T, Williamson A, Lee TS, Petroff OA, de Lanerolle NC. Glutamate and astrocytes-key players in human mesial temporal lobe epilepsy? Epilepsia. 2008;49 Suppl 2:42-52.
28. Ueda Y, Yokoyama H, Nakajima A, Tokumaru J, Doi T, Mitsuyama Y. Glutamate excess and free radical formation during and following kainic acid-induced status epilepticus. Exp Brain Res. 2002;147(2):219-26.
29. Morimoto K, Fahnestock M, Racine RJ. Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog Neurobiol. 2004; 73(1):1-60.
]]>30. Rogawski MA, Kurzman PS, Yamaguchi SI, Li H. Role of AMPA and GluR5 kainate receptors in thedevelopment and expression of amygdala kindling in the mouse. Neuropharmacology. 2001;40(1): 28-35.
31. Deshpande LS, Lou JK, Mian A, Blair RE, Sombati S, Attkisson E, et al. Time course and mechanism of hippocampal neuronal death in an in vitro model of status epilepticus: role of NMDA receptor activation and NMDA dependent calcium entry. Eur J Pharmacol. 2008;583(1):73-83.
32. Rice AC, DeLorenzo RJ. NMDA receptor activation during status epilepticus is required for the development of epilepsy. Brain Res. 1998; 782(1-2):240-7.
33. Ullal G, Fahnestock M, Racine R. Time-dependent effect of kainate-induced seizures on glutamate receptor GluR5, GluR6, and GluR7 mRNA and Protein Expression in rat hippocampus. Epilepsia. 2005;46(5):616-23.
34. Vincent P, Mulle C. Kainate receptors in epilepsy and excitotoxicity. Neuroscience. 2009;158(1):309-23.
]]>35. Lado FA, Laureta EC, Moshe SL. Seizure-induced hippocampal damage in the mature and immature brain. Epileptic Disord. 2002;4(2):83-97.
36. Cherubini E, Ben-Ari Y, Krnjevic K. Anoxia produces smaller changes in synaptic transmission, membrane potential, and input resistance in immature rat hippocampus. J Neurophysiol. 1989;62(4):882-95.
37. Stafstrom CE, Holmes GL. Effects of uncontrolled seizures. Neural changes in animal models. Adv Exp Med Biol. 2002;497:171-94.
38. Marks JD, Friedman JE, Haddad GG. Vulnerability of CA1 neurons to glutamate is developmentally regulated. Brain Res Dev Brain Res. 1996; 97(2):194-206.
39. Albala BJ, Moshe SL, Okada R. Kainic-acid-induced seizures: a developmental study. Brain Res. 1984;315(1):139-48.
]]>40. Bender R, Baram TZ. Do prolonged febrile seizures injury hippocampal neurons? Insights from animal models. In: Baram TZ, Shinnar S. editors. Febrile seizures. San Diego: Academic Press. 2002. p.583-7.
41. Berger ML, Tremblay E, Nitecka L, Ben-Ari Y. Maturation of kainic acid seizure-brain damage syndrome in the rat. III. Postnatal development of kainic acid binding sites in the limbic system. Neuroscience. 1984;13(4):1095-104.
42. Ben-Ari Y, Tremblay E, Riche D, Ghilini G, Naquet R. Electrographic, clinical and pathological alterations following systemic administration of kainic acid, bicuculline or pentetrazole: metabolic mapping using the deoxyglucose method with special reference to the pathology of epilepsy. Neuroscience. 1981;6(7):1361-91.
43. Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14(2):375-403.
44. Schwob JE, Fuller T, Price JL, Olney JW. Widespread patterns of neuronal damage following systemic or intracerebral injections of kainic acid: a histological study. Neuroscience. 1980; 5(6):991-1014.
]]>45. Sloviter RS. The neurobiology of temporal lobe epilepsy: too much information, not enough knowledge. C R Biol. 2005;328(2):143-53.
46. Sperk G, Lassmann H, Baran H, Seitelberger F, Hornykiewicz O. Kainic acid-induced seizures: dose-relationship of behavioural, neurochemical and histopathological changes. Brain Res. 1985;338(2):289-95.
47. Anguelova E, Smirnova T. Differential expression of small heat shock protein 27 in the rat hippocampus and septum after fimbria-fornix lesion. Neurosci Lett. 2000;280(2):99-102.
48. Valentim LM, Geyer AB, Tavares A, Cimarosti H, Worm PV, Rodnight R, et al. Effects of global cerebral ischemia and preconditioning on heat shock protein 27 immunocontent and phosphorylation in rat hippocampus. Neuroscience. 2001;107(1):43-9.
49. Planas AM, Soriano MA, Estrada A, Sanz O, Martin F, Ferrer I. The heat shock stress response after brain lesions: induction of 72 kDa heat shock protein (cell types involved, axonal transport, transcriptional regulation) and protein synthesis inhibition. Prog Neurobiol. 1997;51(6):607-36.
]]>50. Planas AM, Soriano MA, Ferrer I, Rodriguez Farre E. Kainic acid-induced heat shock protein-70, mRNA and protein expression is inhibited by MK-801 in certain rat brain regions. Eur J Neurosci. 1995;7(2):293-304.
51. Yenari MA, Fink SL, Sun GH, Chang LK, Patel MK, Kunis DM, et al. Gene therapy with HSP72 is neuroprotective in rat models of stroke and epilepsy. Ann Neurol. 1998;44(4):584-91.
52. Pozas E, Ballabriga J, Planas AM, Ferrer I. Kainic acid-induced excitotoxicity is associated with a complex c-Fos and c-Jun response which does not preclude either cell death or survival. J Neurobiol. 1997;33(3):232-46.
53. Gass P, Herdegen T. Neuronal expression of AP1 proteins in excitotoxic neurodegenerative disorders and following nerve fiber lesions. Progr Neurobiol. 1995;47(4-5):257-90.
54. Kasof GM, Mandelzys A, Maika SD, Hammer RE, Curran T, Morgan JI. Kainic acid-induced neuronal death is associated with DNA damage and a unique immediate-early gene response in c-fos-lacZ transgenic rats. J Neurosci. 1995;15(6):4238-49.
]]>55. Chen ZL, Strickland S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell. 1997;91(7):917-25.
56. Tan Z, Levid J, Schreiber SS. Increased expression of Fas (CD95/APO-1) in adult rat brain after kainate-induced seizures. Neuroreport. 2001; 12(9):1979-82.
57. Gillardon F, Wickert H, Zimmermann M. Up-regulation of bax and down-regulation of bcl-2 is associated with kainate-induced apoptosis in mouse brain. Neurosci Lett. 1995;192(2): 85-8.
58. Lopez E, Pozas E, Rivera R, Ferrer I. Bcl-2, Bax and Bcl-x expression following kainic acid administration at convulsant doses in the rat. Neuroscience. 1999;91(4):1461-70.
59. Gillardon F, Klimaschewski L, Wickert H, Krajewski S, Reed JC, Zimmermann M. Expression pattern of candidate cell death effector proteins Bax, Bcl-2, Bcl-X, and c-Jun in sensory and motor neurons following sciatic nerve transection in the rat. Brain Res. 1996;739(1-2):244-50.
]]>60. Faherty CJ, Xanthoudakis S, Smeyne RJ. Caspase-3-dependent neuronal death in the hippocampus following kainic acid treatment. Brain Res Mol Brain Res. 1999;70(1):159-63.
61. Ferrer I, Lopez E, Blanco R, Rivera R, Krupinski J, Marti E. Differential c-Fos and caspase expression following kainic acid excitotoxicity. Acta Neuropathol. 2000;99(3):245-56.
62. Henshall DC, Chen J, Simon RP. Involvement of caspase-3-like protease in the mechanism of cell death following focally evoked limbic seizures. J Neurochem. 2000;74(3):1215-23.
63. Kienzler F, Norwood BA, Sloviter RS. Hippocampal injury, atrophy, synaptic reorganization, and epileptogenesis after perforant pathway stimulation-induced status epilepticus in the mouse. J Comp Neurol. 2009;515(2):181-96.
64. Sloviter RS, Zappone CA, Harvey BD, Frotscher M. Kainic acid-induced recurrent mossy fiber innervation of dentate gyrus inhibitory interneurons: possible anatomical substrate of granule cell hyper-inhibition in chronically epileptic rats. J Comp Neurol. 2006;494(6):944-60.
]]>65. Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5(4): 1016-22.
66. Bender R, Dubé C, Baram TZ. Mossy fiber sprouting into the inner molecular layer of the dentate gyrus follows prolonged febrile seizures in immature rat model. Epilepsia. 2000;41(suppl 7):76-9.
67. Bendotti C, Pende M, Samanin R. Expression of GAP-43 in the granule cells of rat hippocampus after seizure-induced sprouting of mossy fibres: in situ hybridization and immunocytochemical studies. Eur J Neurosci. 1994;6(4):509-15.
68. Boschert U, O’Shaughnessy C, Dickinson R, Tessari M, Bendotti C, Catsicas S, et al. Developmental and plasticity-related differential expression of two SNAP-25 isoforms in the rat brain. J Comp Neurol. 1996;367(2):177-93.
69. Geddes JW, Hess EJ, Hart RA, Kesslak JP, Cotman CW, Wilson MC. Lesions of hippocampal circuitry define synaptosomal-associated protein-25 (SNAP-25) as a novel presynaptic marker. Neuroscience. 1990;38(2):515-25.
70. Numakawa T, Matsumoto T, Numakawa Y, Richards M, Yamawaki S, Kunugi H. Protective Action of Neurotrophic Factors and Estrogen against Oxidative Stress-Mediated Neurodegeneration. J Toxicol. 2011;2011:405194.
71. Numakawa T, Suzuki S, Kumamaru E, Adachi N, Richards M, Kunugi H. BDNF function and intracellular signaling in neurons. Histol Histopathol. 2010;25(2):237-58.
72. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495-516.
73. Fujikawa DG, Shinmei SS, Zhao S, Aviles ER, Jr. Caspase-dependent programmed cell death pathways are not activated in generalized seizure-induced neuronal death. Brain Res. 2007;1135(1):206-18.
74. Uysal H, Cevik IU, Soylemezoglu F, Elibol B, Ozdemir YG, Evrenkaya T, et al. Is the cell death in mesial temporal sclerosis apoptotic? Epilepsia. 2003;44(6):778-84.
75. Uysal H, Cevik IU, Soylemezoglu F, Elibol B, Ozdemir YG, Evrenkaya T, et al. Is the cell death in mesial temporal sclerosis apoptotic? Epilepsia. 2003;44(6):778-84.
76. Narkilahti S, Nissinen J, Pitkanen A. Administration of caspase 3 inhibitor during and after status epilepticus in rat: effect on neuronal damage and epileptogenesis. Neuropharmacology. 2003;44(8):1068-88.
77. Pollard H, Charriaut-Marlangue C, Cantagrel S, Represa A, Robain O, Moreau J, et al. Kainate-induced apoptotic cell death in hippocampal neurons. Neuroscience. 1994;63(1):7-18.
78. Sloviter RS, Dean E, Sollas AL, Goodman JH. Apoptosis and necrosis induced in different hippocampal neuron populations by repetitive perforant path stimulation in the rat. J Comp Neurol. 1996;366(3):516-33.
79. Charriaut-Marlangue C, Ben-Ari Y. A cautionary note on the use of the TUNEL stain to determine apoptosis. Neuroreport. 1995;7(1):61-4.
80. Roy M, Hom JJ, Sapolsky RM. HSV-mediated delivery of virally derived anti-apoptotic genes protects the rat hippocampus from damage following excitotoxicity, but not metabolic disruption. Gene Ther. 2002;9(3):214-9.
81. Yamamoto A, Murphy N, Schindler CK, So NK, Stohr S, Taki W, et al. Endoplasmic reticulum stress and apoptosis signaling in human temporal lobe epilepsy. J Neuropathol Exp Neurol. 2006; 65(3):217-25.
82. Schindler CK, Pearson EG, Bonner HP, So NK, Simon RP, Prehn JH, et al. Caspase-3 cleavage and nuclear localization of caspase-activated DNase in human temporal lobe epilepsy. J Cereb Blood Flow Metab. 2006;26(4):583-9.
83. Yamamoto A, Schindler CK, Murphy BM, Bellver-Estelles C, So NK, Taki W, et al. Evidence of tumor necrosis factor receptor 1 signaling in human temporal lobe epilepsy. Exp Neurol. 2006;202(2):410-20.
84. Kilany A, Raouf ER, Gaber AA, Aloush TK, Aref HA, Anwar M, et al. Elevated serum Bcl-2 in children with temporal lobe epilepsy. Seizure. 2012; 21(4):250-3.
85. Sakhi S, Bruce A, Sun N, Tocco G, Baudry M, Schreiber SS. p53 induction is associated with neuronal damage in the central nervous system. Proc Natl Acad Sci USA. 1994;91(16):7525-9.
86. Liu H, Cao Y, Basbaum AI, Mazarati AM, Sankar R, Wasterlain CG. Resistance to excitotoxin-induced seizures and neuronal death in mice lacking the preprotachykinin A gene. Proc Natl Acad Sci USA. 1999;96(21):12096-101.
87. Lopez-Meraz ML, Wasterlain CG, Rocha LL, Allen S, Niquet J. Vulnerability of postnatal hippocampal neurons to seizures varies regionally with their maturational stage. Neurobiol Dis. 2010;37(2):394-402.
88. Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, et al. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J Neurochem. 2001;77(1):220-8.
89. Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J Neurosci. 1996;16(4):1337-45.
90. Zeiss CJ. The apoptosis-necrosis continuum: insights from genetically altered mice. Vet Pathol. 2003;40(5):481-95.
91. Schmechel DE. Apoptosis in neurodegenerative disorders. In: Hannum YA, Boustany RM, editors. Apoptosis in neurobiology. Washington DC: CRC Press; 1999. p. 23-48.
92. Martin LJ. Neuronal cell death in nervous system development, disease, and injury (Review). Int J Mol Med. 2001;7(5):455-78.
93. Lorigados L, Orozco S, Morales L, García I, Estupiñán B, Bender JE, et al. Muerte neuronal en la neocorteza de pacientes con epilepsia del lóbulo temporal resistente a fármacos. Neurología. 2008;23(9):555-65.
94. Lopez J, Gonzalez ME, Lorigados L, Morales L, Riveron G, Bauza JY. Oxidative stress markers in surgically treated patients with refractory epilepsy. Clin Biochem. 2007;40(5-6):292-8.
95. Pitkanen A. Drug-mediated neuroprotection and antiepileptogenesis: animal data. Neurology. 2002;59(9 Suppl 5):S27-33.
Received in November, 2011.
Accepted in August, 2012.
]]>
Lourdes Lorigados. Departamento de Inmunoquímica, Centro Internacional de Restauración Neurológica, Cirén. Ave. 25, No. 15805 e/ 158 y 160 Playa, CP 11300, La Habana, Cuba. E-mail: lourdes.lorigados@infomed.sld.cu.
]]>{kind=link}
{kind=link}