Enfermedad de Charcot-Marie-Tooth en numerosos miembros de una familia
Charcot-Marie-Tooth disease in several members of a family
Dra. Tamara Rubio González, I Al. Lisandra Norbet Vázquez II y Al. Adriana de Paz Rosales II
I Policlínico Docente "Camilo Torres Restrepo", Universidad de Ciencias Médicas, Santiago de Cuba, Cuba. ]]> II Facultad de Medicina No. 1, Universidad de Ciencias Médicas, Santiago de Cuba, Cuba.
RESUMEN
La enfermedad de Charcot-Marie-Tooth es una afección degenerativa del sistema nervioso periférico, que presenta gran heterogeneidad genética y clínica. La presentación con patrón autosómico dominante, conocida en algunas clasificaciones como de tipo 1, es la más frecuente; asimismo, la confección del árbol genealógico resulta ser el instrumento de mayor importancia para conocer el tipo de herencia. A tales efectos, se describen 2 casos clínicos pertenecientes a una familia con 35 miembros afectados por este trastorno neurológico, atendidos en el Centro Provincial de Genética Médica de Santiago de Cuba.
Palabras clave: enfermedad de Charcot-Marie-Tooth, neuropatía sensitiva motora, neuropatía hereditaria mixta.
ABSTRACT
Charcot-Marie-Tooth disease is a degenerative affection of the peripheral nervous system that presents great genetic and clinic heterogeneity. The presentation with autosomal dominant pattern, well-known in some classifications as type I, is the most frequent; also, the making of the genealogical tree turns out to be the most important instrument to know the inheritance type. To such effects, 2 case reports belonging to a family with 35 members affected by this neurological dysfunction are described, assisted in the Provincial Center of Medical Genetics in Santiago de Cuba.
Key words: Charcot-Marie-Tooth disease, motor sensitive neuropathy, mixed hereditary neuropathy.
]]>
INTRODUCCIÓN
La enfermedad de Charcot-Marie-Tooth (CMT) representa una de las neuropatías hereditarias mixtas más comunes, con una incidencia de 1 por cada 2 500 nacidos vivos, según se ha informado en varias latitudes. 1 Es una alteración monogénica con heterogeneidad genética, la cual es responsable de la heterogeneidad clínica observada. La presentación con patrón autosómico dominante, conocida en algunas clasificaciones como de tipo 1, es la más frecuente, con cerca de 80-90 % del total de casos.
Por otra parte, la presentación ligada a X se registra con una frecuencia de 20 % aproximadamente y el patrón de herencia recesivo no llega a 10 %.1 Las manifestaciones clínicas son amplias y no hay una correlación genotipo-fenotipo establecida; sin embargo, algunas características clinicoelectrofisiológicas permiten clasificar este padecimiento. Típicamente, el paciente cursa con atrofia, hiporreflexia y debilidad muscular distal, asociada a pérdida sensorial, que va de leve a moderada.2
En los últimos años se ha avanzado considerablemente en el conocimiento molecular y genético de las alteraciones que se presentan en las neuropatías hereditarias. Por esto, se debe realizar asesoramiento genético a las familias, no sin antes advertir que en este medio existen limitaciones técnicas y económicas que dificultan el acceso a las pruebas requeridas para determinar con exactitud el gen afectado. Lo anterior se agrava si se tiene en cuenta que no siempre se cuenta con el acceso a estudios electrofisiológicos e histopatológicos; no obstante, la utilización del método clínico permite el diagnóstico y determinación de la forma clínica de la enfermedad. Para conocer el tipo de herencia, la confección del árbol genealógico es el instrumento de mayor importancia.
CASOS CLÍNICOS
Caso 1
Se describe el caso clínico de un paciente mestizo de 10 años de edad, nacido de un parto distócico por cesárea, con peso al nacer de 9,2 libras, llanto vigoroso, sin alteraciones neonatales ni antecedentes prenatales de interés, así como también con un desarrollo psicomotor normal, quien desde hacía un año comenzó a presentar cansancio, dolores en miembros inferiores y dificultad para la marcha, por lo cual acudió a la Consulta de Neurología del Hospital Infantil Sur de Santiago de Cuba y luego fue remitido, para su estudio, al Centro Provincial de Genética de Santiago de Cuba en octubre del 2000.
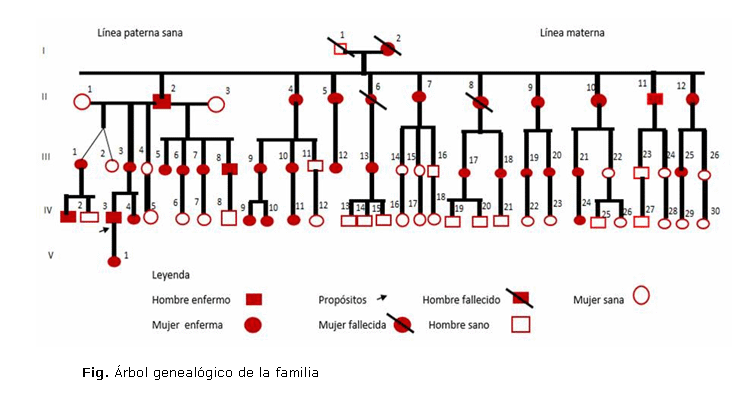
Como antecedentes patológicos familiares por la línea materna figuraron la presencia de polineuropatía periférica en 33 miembros de la familia, documentado en el árbol genealógico confeccionado (figura) y madre con hipotiroidismo; por la línea paterna, asma bronquial.
• Examen físico ]]>
- Cráneo normoconfigurado, orejas grandes y despegadas, nariz ancha con anteversión de las alas y rafe en el filtro nasolabial.- Tórax estrecho, pectus excavatum, panículo adiposo disminuido y escoliosis.
- Ausencia de alteraciones abdominales y genitales.
- Extremidades superiores: limitación bilateral para la extensión de la articulación del codo, más evidente en el izquierdo, así como limitación en la abducción de la muñeca de ambas manos, más acentuada en la izquierda.
- Extremidades inferiores: afinamiento distal de miembros inferiores en forma de "pata de cigüeña" y disminución de la fuerza muscular en ambos miembros.
• Exámenes complementarios
- Estudios de conducción nerviosa: al estimular el nervio tibial posterior bilateralmente se obtuvo retardo de la conducción nerviosa, compatible con daño mielínico severo en fibras sensitivas periféricas y/o centrales, a lo largo de la vía somatosensorial (sistema dorsal-lemniscal).
- Potencial evocado auditivo del tronco cerebral: en el oído derecho se detectó conducción normal en todos sus segmentos; en el izquierdo, retardo en la conducción nerviosa en el segmento retrococlear de la vía auditiva a nivel del tallo encefálico, localizado entre el nervio auditivo (porción intraaxial) y la protuberancia ventrocaudal.
- Potencial evocado visual: normal.
- Electromiografía: patrón de contracción neurógeno periférico en miembro superior derecho, signos de daño mielínico en fibras motoras radiculoplexurales y miembro superior izquierdo normal, igual que los miembros inferiores. ]]>
Caso 2
Se describe el caso clínico de una niña mestiza de 5 años de edad, hija del propósitus descrito anteriormente, producto de un parto eutócico, con peso al nacer de 8,3 libras, puntaje de Apgar 9/9 y sin alteraciones neonatales, que también caminó tardíamente y aún no tenía control de esfínteres, quien fue llevada a consulta porque presentaba dificultad para caminar.
Como antecedente patológico familiar por línea paterna se destacó la presencia de polineuropatía periférica y como antecedentes prenatales: madre con amenaza de aborto y de parto pretérmino, además de moniliasis vaginal durante toda la gestación, de manera que fue tratada con metrodinazol, rocephin y óvulos de clotrimazol en el tercer trimestre; también presentó anemia a los 8 meses de embarazo, por lo que fue transfundida. Cabe agregar que los resultados de los estudios prenatales (alfafetoproteína y ecografías) resultaron normales.
• Examen físico
- Cabeza de forma y tamaño normales, aspecto mofletudo de la cara, presencia de surco infraorbitario bilateral y anteversión de alas nasales, orejas grandes y despegadas.
- Cuello, tórax, abdomen, columna vertebral, piel y genitales: sin rasgos dismórficos significativos.
- Extremidades: se constató hiperextensibilidad bilateral del codo y de las articulaciones interfalángicas, genu recurvatum y ligero afinamiento distal en miembros inferiores.
- Examen neuromuscular: pérdida de la fuerza muscular de todos los miembros, dificultad para subir escaleras, saltar, ponerse de pie y para coger la cuchara. Marcha bamboleante e inteligencia normal.
• Exámenes complementarios ]]>
Estudio de conducción nerviosa (conducción nerviosa sensitiva): daño anormal del nervio mediano derecho e izquierdo. Sural derecho normal. Sural izquierdo con daño axonal. Conducción nerviosa motora: daño axonomielínico en fibras motoras de los nervios medianos derecho e izquierdo, del nervio cubital, del peroneo derecho e izquierdo (músculo pedio), del tibial anterior derecho e izquierdo y del tibial posterior.
COMENTARIOS
Las formas clínicas de Charcot-Marie-Tooth (CMT) incluyen: CMT1, CMT2, CMT3, CMT4, y CMTX,3-5 lo que responde a una gran heterogeneidad genética.
• CMT1: es la más frecuente, resulta de alteraciones en la capa de mielina que recubre los nervios. De esta hay varios tipos: 6-8
- CMT1A: enfermedad autosómica dominante, que resulta de una duplicación del gen PMP-22, con locus 17p11.2-12, que codifica la proteína 22, componente fundamental de la capa de mielina; también se registra con menos frecuencia tetrasomía y mutaciones puntuales.
- CMT1B: enfermedad autosómica dominante causada por mutaciones del gen MPZ, con locus 1q21-23, que codifica la proteína cero (P0), otro componente de la envoltura de mielina.
- CMT1C: se produce por mutaciones en el gen simple, que codifica la proteína LITAF y tiene que ver con la estimulación de monocitos y macrófagos.
- CMT1D: producida por mutaciones del gen EGR2, que regula la expresión de genes fundamentales para la mielinización como MPZ, PMP-22, GJB1 y periaxina.
- CMT1F: producido por mutación del gen NEFL, que codifica la cadena ligera de los neurofilamentos. ]]>
• CMT2: se debe a anormalidades en el axón de la célula nerviosa periférica y no de la envoltura de mielina, por lo que en el electromiograma no se dañan tanto las velocidades de conducción. La edad de inicio es posterior, la escoliosis o pies cavos son menos frecuentes y graves y los reflejos se conservan por más tiempo. En la mayoría de las series descritas, CMT2 es menos común que CMT1.1,4• CMT3 o Dejerine-Sottas: neuropatía desmielinizante grave de inicio en la infancia. Los lactantes padecen de atrofias y debilidades musculares graves y problemas sensoriales. Se debe a una mutación específica puntual de los genes P0 o PMP-22.
• CMT4: abarca varios subtipos de neuropatías desmielinizantes autosómicas recesivas motoras y sensoriales. El defecto genético aún no ha sido identificado.
• CMTX: enfermedad dominante ligada al cromosoma X, causada por una mutación en el gen conexina 32, localizado en Xq13-1. La proteína conexina 32 se presenta en las células de Schwann, que recubren los axones del nervio, de manera que constituye un solo segmento de la capa de mielina. Los varones que heredan un gen mutado de sus madres presentan síntomas de moderados a graves al final de la niñez o en la adolescencia; en las hembras también aparecen algunos, por inactivación del cromosoma X con el alelo normal.3,7,8
Las principales características del síndrome de Charcot-Marie-Tooth se deben a las alteraciones en la neurona motora inferior acompañada de signos sensoriales, que en combinación ocasionan una neuropatía neurosensorial. En el fenotipo clásico, los síntomas inician en la primera o segunda décadas de la vida, con curso lento y progresivo (debilidad y atrofia muscular con hiporreflexia o arreflexia en extremidades inferiores).
Posteriormente, por la cronicidad de la neuropatía motora, puede aparecer pie cavo, dedos en martillo, atrofia muscular en piernas y tercio inferior de muslos, lo cual da lugar a las "patas de cigüeña" o de "botella de champán invertida", debido a la pérdida de masa muscular; asimismo, puede presentarse dificultad para caminar y correr, marcha equina, disminución de la percepción del dolor, temperatura o propiocepción en miembros inferiores, manos en forma de garra, temblor en manos, calambres, pies fríos, hiperqueratosis plantar y acrocianosis, aunque ciertas características clínicas son difícilmente distinguibles entre las formas axonal y desmielinizante.
Los cambios electrofisiológicos son detectables en la infancia, con una prolongación de la latencia motora distal en los primeros meses de vida y, más tarde, alteración en la velocidad de conducción nerviosa. La mayoría de los pacientes no se quejan de síntomas sensitivos, de modo que la expresión más patente de la alteración de la sensibilidad es la inestabilidad. El dolor espontáneo no es frecuente, pero sí lo es el dolor en los pies ocasionado por el mal apoyo, por callosidades o deformidades.9,10
Según los hallazgos del estudio electrofisiológico, la enfermedad se clasifica en 3 tipos a saber:7,8
- Tipo 1 (Charcot Marie Tooth 1 o desmielinizante): la velocidad de conducción motora de los nervios está ralentizada, generalmente inferior a 38 m/s. Existen alteraciones motoras y sensitivas.
- Tipo 2 (Charcot Marie Tooth 2 o forma axonal): la velocidad de conducción está normal o casi normal, generalmente más de 38 m/s. Hay alteraciones motoras y sensitivas. ]]>
- Neuropatía motora hereditaria (NMH) o atrofia espinal distal (AED): la velocidad de conducción es normal. En esta forma no hay alteraciones sensitivas en el estudio electrofisiológico, o sea, la sensibilidad está preservada.Los 35 integrantes de esta familia padecen una neuropatía hereditaria sensitivo-motora (NHSM) o enfermedad de Charcot-Marie-Tooth, con debilidad y degeneración musculares y alguna pérdida de sensibilidad en los pies, la parte inferior de las piernas, las manos y los antebrazos. Cabe agregar que como principales características se destacan: inicio en la primera década de la vida, curso lento pero progresivo de la enfermedad, debilidad en miembros inferiores, atrofia muscular distal, pie cavo (según pasa el tiempo) y deformidad de las piernas, como "botella de champán invertida".
De acuerdo con los estudios de conducción periférica, los pacientes mostraron retardo de la conducción nerviosa, compatible con daño mielínico severo en fibras sensitivas periféricas y/o centrales, a lo largo de la vía somatosensorial (sistema dorsal-lemniscal). Teniendo en cuenta todos los aspectos clínicos y electrofisiológicos mencionados, así como el patrón de herencia autosómico dominante mostrado en esta familia se concluye que se trata de la enfermedad de Charcot-Marie-Tooth con forma clínica CMT1A.
REFERENCIAS BIBLIOGRÁFICAS
1. Lara Aguilar RA, Juárez Vázquez J, Juárez Rendón K, Gutiérrez Amavizca J, Barros Núñez P. Enfermedad de Charcot-Marie-Tooth: actualidad y perspectivas. Arch Neurocien (Mex). 2012;17(2):110-8.
2. Reilly MM, Shy ME, Muntoni F, Pareyson D. 168th ENMC International Workshop: outcome measures and clinical trials in Charcot-Marie-Tooth disease (CMT). Neuromuscul Disord. 2010;20(12):839-46.
3. Katitji B, Koontz D. Disorders of the peripheral nerves. In: Daroff RB, Fenichel GM, Jankovic J, Mazziotta JC. Bradley's Neurology in Clinical Practice. 6th. Philadelphia: Elsevier Saunders;2012.
4. Sevilla T, Vilchez J. Diferentes fenotipos del síndrome de Charcot-Marie-Tooth causados por mutaciones del mismo gen: ¿siguen siendo útiles los criterios de clasificación clásicos? Neurol. 2004;19(5):264-71.
5. Pareyson D, Reilly MM, Schenone A, Fabrizi GM, Cavallaro T, Santoro L, et al. Ascorbic acid in Charcot-Marie-Tooth disease type 1A (CMT-TRIAAL and CMT-TRAUK): a double-blind randomised trial. Lancet Neurol. 2011;10(4):320-8.
6. Shy ME. Peripheral neuropathies. In: Goldman L, Schafer AI. Goldman's Cecil Medicine. 24th. Philadelphia: Elsevier Saunders; 2011.
7. Herman M, Delmis J, Ivaniseviv M, Zupic T. Pregnancies and deliveries in patients with Charcot-Marie-Tooth disease. Acta Med Croatica. 2010;64(3):215-20.
8. Hernández Zamora E, Arenas Sordo ML. El diagnóstico de las neuropatías periféricas hereditarias y la genética molecular. Acta Ortop Mex. 2008;22(4):268-77.
9. Bird TD. Charcot-Marie-Tooth hereditar y neuropathy over view. In: Pagon RA, Bird TD, Dolan CR, Stephens K. Gene Reviews. Seattle: University of Washington; 2011.
10. Castañeda Fernández JA, del Corral García J. Neuropatía periférica. MEDISAN. 2003 [citado 15 Mar 2015];7(4). Disponible en: http://bvs.sld.cu/revistas/san/vol7_4_03 /san07403.htm
Recibido: 26 de junio de 2015.
Aprobado: 19 de diciembre de 2015.
]]> Tamara Rubio González. Policlínico Docente "Camilo Torres Restrepo", calle Heredia nr 354, entre Calvario y Reloj, Santiago de Cuba, Cuba. Correo electrónico:trubio@medired.scu.sld.cu ]]>
{kind=link}