Manifestaciones musculoesqueléticas de la enfermedad de Fabry: reporte de un caso
Musculoskeletal manifestations of Fabry´s disease: a case report
Blanca Elena Ríos Gomes Bica I, Lina Maria Saldarriaga Rivera II, Breno Valdetaro Bianchi III
I Msc. PhD. Especialista en Reumatología Pediátrica. Departamento de Reumatología del Hospital Universitario Clementino Fraga Filho. Universidad Federal de Rio de Janeiro, Brasil.
II Especialista en Medicina Interna y Reumatología. Departamento de Reumatología del Hospital Universitario Clementino Fraga Filho. Universidad Federal de Rio de Janeiro, Brasil. ]]>
III Especialista en Medicina Interna y Reumatología. Departamento de Reumatología del Hospital Universitario Clementino Fraga Filho. Universidad Federal de Rio de Janeiro, Brasil.
RESUMEN
La enfermedad de Anderson-Fabry es una enfermedad genética, de carácter hereditario, que causa la deficiencia o ausencia de la enzima alfa-galactosidasa llevando al acúmulo de globotriacilceramida en diversas células causando sus manifestaciones clínicas, siendo considerada la segunda enfermedad de depósito mas común después de la enfermedad de Gaucher. Describimos el caso de un paciente que presentó manifestaciones musculoesqueléticas compatibles con la enfermedad de Fabry.
Palabras clave: Enfermedad de Fabry, manifestaciones musculoesqueléticas, genética.
ABSTRACT
Anderson-Fabry´s disease is a genetic and hereditary disorder caused by the deficiency or absence of the enzyme alpha-galactosidase leading to accumulation of globotriacilceramida in different cells causing many clinical manifestations. It is considered the second most common storage disease after Gaucher´s disease. We describe a patient who presented musculoskeletal manifestations of Fabry´s disease.
Keywords: Fabry´s disease, musculoskeletal manifestations, genetics.
]]>
INTRODUCCIÓN
La enfermedad de Fabry (EF) también conocida como enfermedad de Anderson-Fabry o angiokeratoma corporis fue descrita de manera independiente y casi simultánea por los dermatólogos William Anderson en Inglaterra y Johannes Fabry en Alemania, en 1898.1
Es una de las 45 enfermedades de depósito lisosómico, hereditaria y está ligada al cromosoma X, presentando un error en el metabolismo de los glicoesfingolipidos debido a la deficiencia de la enzima galactosidasa A (GAL) que lleva al acúmulo de globotriacilceramida (GL-3) en diversas células, causando sus manifestaciones clínicas. La α-GAL es codificada por un gen denominado GALA de 12-Kb situado en el brazo largo del cromosoma X (región Xq22.1) que compromete cerca de 7 exones que varían entre 92 y 291 pares de bases.2
El defecto genético que produce la enfermedad es extremamente heterogéneo y hasta el momento ya fueron descritas más de 300 mutaciones. La mayoría de las familias de los pacientes poseen mutaciones específicas para cada uno de sus componentes.3
La EF es considerada la segunda enfermedad de depósito más común después de la enfermedad de Gaucher con una prevalencia estimada entre 1:17.000 a 1:117.000 personas.4 Su incidencia es mas común en individuos caucásicos, pero puede afectar a cualquier raza. La incidencia en mujeres es subestimada debido al amplio espectro de manifestaciones clínicas pudiendo ser desde asintomática o tan grave como en los hombres.
Muchos pacientes con la EF son diagnosticados incorrectamente o tardíamente habiendo consultado diferentes especialistas antes de la obtención de un diagnóstico preciso.
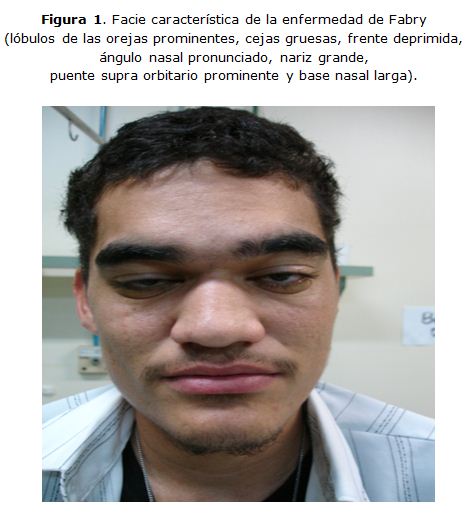
Los pacientes con la forma clásica de la enfermedad presentan fenotipos semejantes, con facie característica (lóbulos de las orejas prominentes, cejas gruesas, frente deprimida, ángulo nasal pronunciado, nariz grande, puente supra orbitario prominente y base nasal larga).
Las manifestaciones iniciales más frecuentes son generalmente dermatológicas, neurológicas y gastrointestinales. Las manifestaciones musculoesqueléticas son raras pudiendo ocurrir osteoporosis, osteonecrosis o artropatía neuropática.5
]]> PRESENTACIÓN DEL CASO
Paciente masculino de 23 años, blanco, soltero, natural de Rio de Janeiro. Remitido al servicio de reumatología después de 15 años de iniciado los síntomas. Presentaba crisis de acroparestesias en miembros superiores e inferiores con duración de semanas a meses desde los 8 años de edad, en ocasiones con necesidad de hospitalizaciones para analgesia.
Estas crisis eran acompañadas de fiebre de hasta 39 °C y angioqueratomas palmoplantares. A los 16 años de edad presentó amaurosis del ojo izquierdo sin diagnóstico etiológico y también taquiarritmia sin otras investigaciones etiológicas.
Debido a esas crisis frecuentes el paciente dejó los estudios a los 13 años de edad y jamás trabajó, presentando trastornos afectivos y de comportamiento depresivo.
Ningún miembro de su familia presentaba enfermedad de Fabry, insuficiencia renal o cardíaca de causa conocida.
Al examen físico presentaba facie característica de la EF (lóbulos de las orejas prominentes, cejas gruesas, frente deprimida, ángulo nasal pronunciado, nariz grande, puente supraorbitario prominente y base nasal larga. [Figura 1]
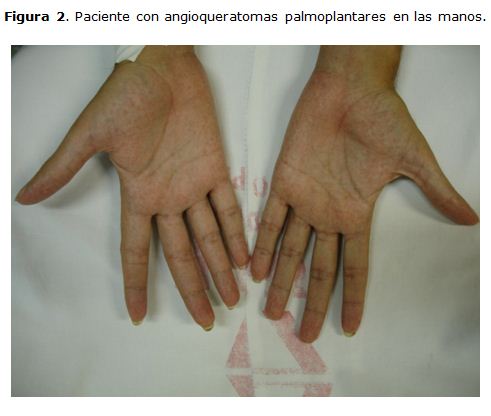
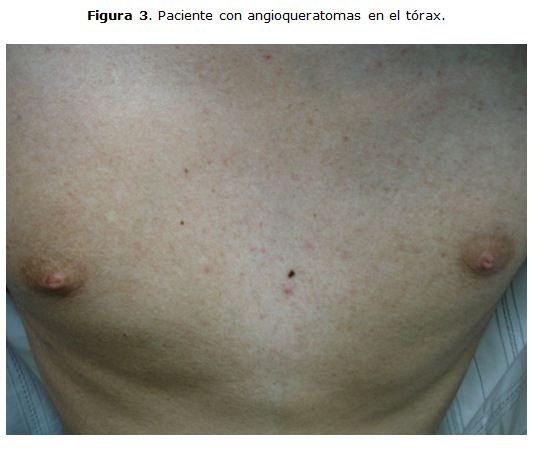
Lesiones eritematosas puntiformes palmoplantares que desaparecían a la digito presión, angioqueratomas en el tórax, e hipocratismo digital. [Figura 2] y [Figura 3]. También presentaba artritis en rodillas, asociado a edema y con limitación para la movilidad, sin manifestaciones neurológicas. Se le realizó una evaluación oftalmológica la cual evidencio córnea verticillata.
]]> El paciente presentaba exámenes recientes de función hepática, renal, bioquímica, hemograma, reactantes de fase aguda, factor reumatoide, prueba de Coombs, perfil tiroideo, radiografía de tórax, electrocardiograma y ecocardiograma sin alteraciones.Presentaba anticuerpos antinucleares 1/80 con patrón nuclear punteado fino. Radiografía de manos y pies con reducción difusa de la densidad ósea, radiografía de rodillas bilateral con aumento de tejidos blandos, osteopenia, sin disminución del espacio articular. Densitometría ósea con datos de osteopenia. Se sospechó de enfermedad de Fabry siendo realizada niveles séricos de α-GAL reportando un valor de 2,4 μmol/L/h (Valor de Referencia >2μmol/L/h). En razón de la fuerte sospecha diagnóstica fue solicitado niveles leucocitarios de α-GAL reportando < 0,6 nmol/L/h (Valor de Referencia 30-63 nmol/L/h). El paciente fue remitido al departamento de genética, donde fue realizado el perfil genético, encontrándose una mutación (c.801+3A>T) en 5 intrones del gen GALA que nunca había sido descrita anteriormente en la EF. Ese hallazgo confirmó el gran polimorfismo encontrado en esa enfermedad.
El paciente fue tratado inicialmente con prednisona 20 mg/día con reducción gradual hasta su retirada, presentando mejoría del cuadro articular y posteriormente una vez conocido el diagnóstico se le suministró terapia de reposición enzimática a una dosis de 3 frascos de 50 mg IV cada 2 semanas, presentando mejoría de los síntomas.
DISCUSIÓN
La EF es una enfermedad rara, con compromiso sistémico, que lleva a insuficiencia renal, insuficiencia cardíaca y aumento de la mortalidad cardiovascular cuando es diagnosticada y tratada tardíamente. El tiempo medio para el diagnóstico es de 13.7 a 16.3 años lo que conlleva aumentar la tasa de mortalidad.6 En nuestro paciente el tiempo para el diagnóstico fue de 18 años.
El diagnóstico es confirmado con los niveles séricos de GAL donde, los niveles séricos sirven como test de screening teniendo menor sensibilidad que los niveles leucocitarios. La confirmación genética no es necesaria en los pacientes del sexo masculino, pero en caso de duda debe ser realizada.7
Después del diagnóstico de EF es necesaria la investigación de los familiares, ya que en el sexo femenino la enfermedad puede presentarse oligosintomática. A pesar de no existir antecedentes familiares de enfermedad renal crónica, miocárdica o cerebrovascular en nuestro paciente, fue realizada investigación genética a los familiares, donde encontramos una mutación heterocigótica (c.801+3A>T) en la madre del paciente.
Las manifestaciones clínicas de la EF son causadas básicamente por acúmulo de GB-3 en los tejidos y endotelio causando los síntomas referidos por el paciente. La fiebre normalmente está relacionada a perdida de control térmico causado por la hipo o anhidrosis. A pesar de que el paciente no refirió tal fenómeno probablemente pudo ser el responsable de la fiebre encontrada en las crisis.
La etiología de las acroparestesias se debe al depósito de GB-3 en la vasa nervorum y se inicia en las primeras décadas de la enfermedad, pudiendo ser precipitadas por el ejercicio físico, calor o cambios de temperatura. A lo largo de los años esas crisis de dolor tienden a desaparecer por la destrucción completa de los ejes nerviosos. En el paciente descrito, las crisis de dolor conllevaron a presentar alteraciones psicológicas y sociales. En casos extremos son descritas hasta tentativas de suicidios debido a las crisis dolorosas.8
]]> La córnea verticillata está presente en casi todos los pacientes homocigotos y en 70 a 90 % de los heterocigotos.Otros hallazgos oftalmológicos son tortuosidades de vasos conjuntivales y de la retina. Neuropatía óptica isquémica y oclusión de la arterial central de retina son manifestaciones raras y que pueden justificar el cuadro de amaurosis del paciente citado.9
En individuos heterocigóticos la manifestación renal inicial es isostenuria y proteinuria sea de origen glomerular o tubular, que inician en el final de la adolescencia o el inicio da 3ª década de vida.10 En nuestro paciente no encontramos evidencia de daño renal.
El compromiso cardiovascular es más tardío, generalmente encontrado en la 5ª década de la vida con hipertrofia ventricular concéntrica y la enfermedad cerebrovascular.
Disturbios de la conducción también pueden ser encontrados en este rango de edad.11
La taquiarritmia presentada por el paciente fue a los 16 años de edad. Al momento de su diagnóstico no observamos alteraciones estructurales en el ecocardiograma y su electrocardiograma estaba normal.
El principal compromiso músculo esquelético reportado es la osteoporosis. Su etiología puede ser multifactorial, como el uso de medicamentos que alteran el metabolismo óseo como la carbamazepina, disturbio de absorción intestinal de 25-hidroxivitamina D. También puede ser encontrado osteonecrosis o artropatía neuropática debido al compromiso vascular y neuropático respectivamente.12
Nuestro paciente presentó datos de osteopenia demostrado en la densitometría ósea de manos, pies y rodillas.
Presentó además un cuadro de artritis de rodillas, la cual mejoró con dosis de 20 mg/día de prednisona, sin manifestaciones neuropáticas.
Solo la terapia de reposición enzimática cada 2 semanas, mejoró el resto de los síntomas que presentaba.
]]>CONCLUSIONES
A pesar del cuadro clínico de la EF tener algunas peculiaridades que tornan el diagnóstico clínico simple, la variedad de presentaciones de la enfermedad y el desconocimiento de su patología y manifestaciones clínicas por los profesionales de la salud retrasan el diagnóstico y por consiguiente su tratamiento oportuno, aumentando la morbimortalidad y empeorando la calidad de vida del paciente con EF.
REFERENCIAS BIBLIOGRÁFICAS
1. Boggio P, Luna PC, Abad ME, Larralde M, Fabry disease, An Bras Dermatol. 2009;84(4):367-76.
2. Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry's disease. Ceramidetrihexosidase deficiency. N Engl J Med. 1967; 276(21):1163-67.
3. Hauser AC, Lorenz M, Sunder-Plassmann G. The expanding clinical spectrum of Anderson-Fabry disease: a challenge to diagnosis in the novel era of enzyme replacement therapy, J Int Medicine 2004;255(6):629-36.
4. Germain DP. Fabry disease. Orphanet J Rare Dis 2010; 5:30.
5. Germain DP, Benistan K, Boutouyrie P, Mutschler C. Osteopenia and osteoporosis: previously unrecognized manifestations of Fabry disease. Clin Genet 2005;68:93–5.
]]> 6. Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004;34(3):236-42.7. Garman SC, Garboczi DN. The molecular defect leading to Fabry disease: structure of human alpha-galactosidase. J Mol Biol 2004; 337(2):319-35.
8. Martins AM, Almeida V, Kyosen AO, Takata ET, Delgado AG, Gonçalves AMBF, et al. Guidelines to Diagnosis and monitoring of Fabry Disease and Review of Treatment Experiences, The J of Pediatrics. 2009;155(4):19-31.
9. Desnick RJ, Brady R, Barranger J, Collins AJ, Germain DP, Goldman M, Wilcox WR. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003;138(4):338-46.
10. Alroy J, Sabnis S, Kopp JB. Renal pathology in Fabry disease. J Am Soc Nephrol 2002;13 Suppl 2):S134-8.
11. MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. 2001;38(11):750-60.
12. Sacre K, Lidove O, Leprieur BG, Ouali N, Laganier J, Caillaud C, Papo T, Bone and joint involviment in fabry disease, Scand J Rheumatol. 2010;39(2):171-4.
Los autores refieren no tener conflicto de intereses ]]> Fuente de financiación: ninguna
Recibido: 8 de julio de 2014
Aprobado: 15 de agosto de 2014
Publicado: 1ro de noviembre de 2014
Responsable para la correspondencia Dra. Lina María Saldarriaga Rivera. E-mail: linamarias7@hotmail.com
Rua Professor Rodolpho Paulo Rocco 225 – Ilha do Fundão. Prédio Hospital Universitário Clementino Fraga Filho 9º andar, Cida de Universitária. Rio de Janeiro. CEP: 21944-970, Tel.+55 21 25622723, +55 21 25622266.