










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Obstetricia y Ginecología
versión On-line ISSN 1561-3062
Rev Cubana Obstet Ginecol vol.43 no.4 Ciudad de la Habana oct.-dic. 2017
PRESENTACIÓN DE CASOS
Síndrome de Meckel Gruber en recién nacido indígena sin control prenatal
Meckel Gruber Syndrome in an Indigenous Newborn with No Prenatal Control
Alex Fabián Villa Quigüirí,I Carlos Gafas González,II Anabela del Rosario Criollo Criollo, III Yosbanys Roque HerreraII
I Servicio de Ginecología y Obstetricia. Hospital José María Velasco Ibarra. Tena. Napo. Ecuador.
II Universidad Nacional de Chimborazo, Riobamba, Chimborazo, Ecuador.
III Hospital Provincial General Docente de Riobamba, Chimborazo. Ecuador.
RESUMEN
En el Hospital "José María Velasco Ibarra" de la ciudad del Tena, provincia de Napo, en la República de Ecuador, se reportó el caso de un neonato con Síndrome de Meckel Gruber, de sexo femenino, fruto de un embarazo sin seguimiento prenatal, hijo de padres indígenas no consanguíneos, provenientes de la comunidad amazónica Tamiahurco ubicada en Misahuallí. En este recién nacido se presentó la triada diagnóstica al respecto: riñones hiperplásicos poliquísticos, encefalocele occipital y polidactilia postaxial bilateral, determina el diagnóstico de certeza de esta enfermedad, en la que al menos dos de estos elementos deben estar presentes. Además, puede cursar con malformaciones a nivel oral, genital, del Sistema Nervioso Central (SNC) y fibrosis hepática.
Palabras clave: síndrome de Meckel-Gruber; enfermedades congénitas; control prenatal.
ABSTRACT
At José María Velasco Ibarra Hospital in Tena, Napo province, in the Republic of Ecuador, the case of a female neonate with Meckel Gruber Syndrome is reported. This infant is the result of a pregnancy with no prenatal follow-up, non-consanguineous indigenous parents, from the Tamiahurco Amazon community, in Misahuallí. This newborn had the diagnostic triad of polycystic hyperplastic kidneys, occipital encephalocele and bilateral postaxial polydactyly, which determined this disease diagnosis of certainty, in which at least two of these elements must be present. Other symptoms are oral, genital malformations, hepatic fibrosis and malformations of the Central Nervous System (CNS).
Keywords: Meckel-Gruber syndrome; congenital diseases; prenatal control.
INTRODUCCIÓN
El origen del estudio del Síndrome de Meckel Gruber (SMK) abarca las aportaciones de Friedrich Meckel, George B. Gruber y Opitz y Howe. El primero describió en el año 1822 la presencia en dos hermanos de meningocele occipital, riñones poliquísticos, polidactilia, microcefalia y paladar hendido. Posteriormente, en 1934, el segundo de estos investigadores informó casos similares a los que nombró con el término dicencefalia esplacno-quística y sugirió el origen genético de esta afección. Treinta y cinco años más tarde, en 1969, el tercer equipo investigativo propuso el actual nombre que hoy recibe esta enfermedad, en honor a los precursores de este hallazgo científico.1,2
Esta entidad enmarcada en las del tipo de las ciliopatías, se caracteriza por un desorden autosómico recesivo, de mortalidad absoluta en la totalidad de los casos reportados; con una tasa de prevalencia históricamente descrita, la que varía en un rango entre 1:13 250 y 1:14 0000 nacidos vivos, afectando a individuos de toda raza y etnia, de ambos sexos.3-4
La presencia de la triada: riñones hiperplásicos poliquísticos, encefalocele occipital y polidactilia postaxial bilateral, determina el diagnóstico de certeza de esta enfermedad, en la que al menos dos de estos elementos deben estar presentes; la que a demás pues cursar con malformaciones a nivel oral, genital, del Sistema Nervioso Central (SNC) y fibrosis hepática.5
Al sistematizar la bibliografía especializada6-8 puede observarse que diferentes autores coinciden en que los criterios diagnósticos del SMK están dados por la presencia de al menos dos de las tres características clásicas antes descritas. Además, ante la asociación de una anomalía mayor y dos anormalidades relevantes, se describe otra posibilidad diagnóstica relativa a la patogénesis y manejo de múltiples mutaciones genéticas, lo que genera una variabilidad de formas de presentación de este síndrome, las que se relacionan con alteraciones presentes en diferentes cromosomas. Entre estas combinaciones se encuentran: 17q21-24 al SMK uno (SMK1), 11q13 al SMK dos (SMK2), 8q21.3q22.1 al SMK tres (SMK3), 12q21.31q al SMK cuatro (SMK4), 16q12.2 al SMK cinco (SMK5), y el 4p15.3 al SMK seis (SMK6).
La utilidad clínica del estudio de casos del SMK radica en la necesidad de su detección durante el período prenatal, especialmente en gestantes con antecedentes de anomalías fetales en embarazos previos, debido al alto riesgo de recidiva reportado.5
En tal sentido, la ecografía en el segundo y tercer trimestre del embarazo resulta el mejor método para ese diagnóstico. Sin embargo, autores como Audifred y otros,5 plantean que entre las semanas 11 y 14 los exámenes ultrasonográficos sistemáticos del cráneo, cerebro, riñones, vejiga, manos y pies fetales pueden revelar los primeros hallazgos patológicos.
A la luz de la relación de subordinación e interdependencia que subyace entre los elementos teóricos anteriormente expuestos y la infrecuencia reportada de esta enfermedad en población indígena sin seguimiento prenatal, los autores infieren que dadas las características clínicas presentes en este estudio, el mismo amerita ser compartido con la comunidad científica.
PRESENTACIÓN DE CASO
En el Hospital José María Velasco Ibarra de la ciudad del Tena, provincia de Napo, en la República de Ecuador, se reporta el caso de un neonato de sexo femenino, fruto de un embarazo sin seguimiento prenatal, hijo de padres indígenas no consanguíneos, provenientes de la comunidad amazónica Tamiahurco, ubicada en Misahuallí.
Antecedentes obstétricos
Madre de 33 años de edad, con cinco embarazos anteriores de la misma pareja sexual, de los cuales abortó el tercero a las 12 semanas de gestación sin causa aparente; mientras que los cuatro hijos restantes fueron obtenidos por partos eutócicos. No se recoge información sobre el consumo de medicamentos durante los períodos gestacionales, ni exposición a sustancias tóxicas; así como no existen registros de controles prenatales.
Al examen físico prenatal se evidenció feto único, en situación transversa, con dorso en posición anterior y movimientos presentes; frecuencia cardiaca de 135 a 140 latidos por minuto y ruidos cardiacos débiles según monitoreo fetal. La ecografía obstétrica revela presencia de anencefalia y malformaciones múltiples.
El recién nacido del caso que se presenta es producto de un embarazo pos término, de 43,2 semanas de edad gestacional según fecha de última menstruación, distocia de presentación y ausencia de actividad uterina que motivó la decisión de realizar cesárea de emergencia, obteniéndose un neonato con las características siguientes:
- Estado al nacer: vivo.
- Sexo: femenino.
- Peso: 2920 gr.
- Talla: 40 cm de longitud.
- Perímetro cefálico: 28,5 cm.
- APGAR al primer minuto: 1
- APGAR al quinto minuto:1
- Edad gestacional según el test de Capurro: 42,6 semanas.
- Piel: pálida, hipertricosis leve generalizada y recubierta con vérmix caseoso abundante.
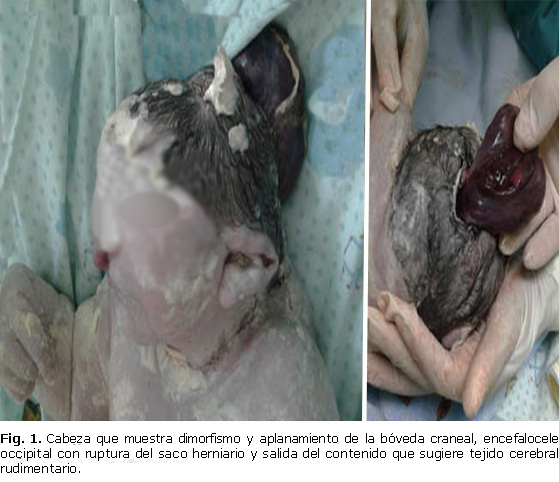
- Cabeza: dimorfismo y aplanamiento de la bóveda craneal, encefalocele occipital con ruptura del saco herniario y salida de contenido que sugiere tejido cerebral rudimentario de color pardo oscuro (Fig. 1), de aproximadamente 10 x 5 x 3 cm de diámetro, con una superficie lisa y brillante, facies aplanada. En región ocular se observó anoftalmia, hipertelorismo, nariz con puente nasal ancho, cartílago nasal aplanado, narinas prominentes. En boca y cavidad oral la existencia de labio leporino, paladar hendido y micrognatia. Orejas de implantación baja, asimétricas y displasias.
- Cuello: corto, ancho y flácido.
- Tórax: ancho y simétrico.
- Aparato respiratorio: murmullo vesicular disminuido.
- Aparato cardiovascular: corazón hipo fonético, bradicardia (50 lpm), saturación de oxígeno al ambiente 50 %.
- Abdomen: simétrico, suave, depresible, ruidos hidroaéreos ausentes, cordón umbilical clampeado donde se observan dos arterias y una vena.
- Sistema reproductor: genitales femeninos normales.
- Ano: permeable.
- Extremidades superiores: manos con polidactilia postaxial o ulnar bilateral (Fig. 2).
- Miembros inferiores: tibias bilaterales en varas. Pie equino varo y polidactilia bilateral (Fig. 2).

El estudio posmortem de Tomografía Axial Computarizado (TAC) simple y de reconstrucciones tridimensionales, mostró:
- Cabeza: cráneo con imagen clásica que refleja defecto óseo y agujero en la escama occipital.
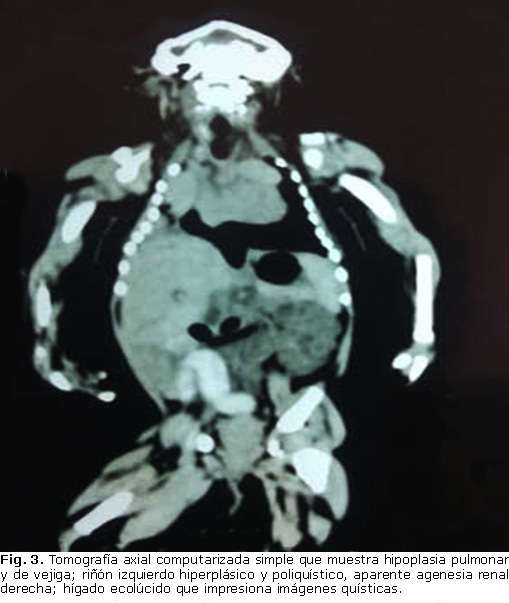
- Pulmones: hipoplasia (Fig. 3).
- Riñones: izquierdo hiperplásico y poliquístico; aparente agenesia renal derecha (Fig. 3).
- Hígado: ecolúcido con patrón ecográfico que impresiona imágenes quísticas (Fig. 3).
- Vejiga: hipoplasia (Fig. 3).
- Miembros inferiores: con incurvación en los huesos largos.
DISCUSIÓN
La literatura contemporánea especializada evidencia que el SMK se enmarca en las enfermedades autosómicas recesiva, en la cual existe el riesgo de que los padres portadores de ese patrón hereditario tengan un hijo afectado en alrededor del 25 % de los embarazos.1,2,5,9
En este estudio, tomando en consideración el historial obstétrico antes descrito, el neonato portador de esa enfermedad representó 20 % de los hijos nacidos vivos de esa pareja sexual.
Para autores como Audifred Salomón, Hernández y otros,1,5 la consanguinidad es considerada como un factor de riesgo de esta enfermedad; sin embargo, esta característica no se observó en el caso que se analiza, atendiendo a la declaración de los padres.
A pesar de que la lógica de la realización y descripción del examen físico se debe realizar en sentido céfalo caudal. A continuación, se expone el análisis de este teniendo en cuenta la relevancia de los elementos que conforman la triada del SMK, los cuales estuvieron presentes en el neonato del caso que se presenta (Fig. 1, 2, 3).
En relación con la displasia renal poliquística, algunos autores 10 coinciden en la hipótesis fisiopatológica de que esta pudiera estar relacionada con un defecto en la interacción entre el blastema y los canales de metanefros, lo que resulta en una anomalía en el desarrollo de la unidad estructural y funcional del riñón (nefronas); convirtiéndose esta, en una de las alteraciones que caracterizan al SMK. Así mismo, la división anárquica de los canales de metanefros da lugar a la aparición de quistes, responsables de la hipertrofia renal. A la luz de este posicionamiento teórico, la realización del estudio anátomo-patológico post-mortem del recién nacido hubiera aportado mayor evidencia sobre este particular; pero debido a que los padres no ofrecieron su consentimiento para la autopsia, esto no fue posible.
La presencia de encefalocele occipital constituye el segundo criterio tomado en consideración para establecer el diagnóstico de certeza del SMK, el que puede aparecer hasta en 90 % de los casos, según reportes de investigadores. Esta malformación se caracteriza por una deficiencia en el cierre del tubo neural a nivel de la región 4; la cual conduce a la ausencia total o parcial de hueso occipital.11,12
El tercer elemento de la triada del SMK está dado por la presencia de polidactilia postaxial bilateral, anomalía que se ataña a la existencia de zonas adicionales de muerte celular interdigital; característica que aparece con una frecuencia menor del 80 % en los casos reportados; sin embargo, cuando esta se manifiesta, afecta a las cuatro extremidades.11,13
La madre de este neonato portador del SMK, no reportó estudios prenatales, hecho que favoreció la detección tardía de esta enfermedad incompatible con la vida fetal; la que, además puso en peligro la salud materna.
En relación con los estudios prenatales de afecciones genéticas, a pesar de que este constituye un indicador evaluativo de la salud fetal y predictor de la calidad de vida de la madre y su producto de la concepción,14 la realización de estos está indisolublemente ligado a factores de orden personal, cultural, económico y político, los que en el caso que se presenta, limitaron su ejecución.
Estudios publicados por Hernández y otros,1 demuestran que la determinación de alfafetoproteína, acetilcolinesterasa, gonadotropina coriónica y fosfatasa alcalina, entre otros, resultan de interés en la detección temprana de enfermedades de origen genético; las que debido a los factores antes referidos no se ofrecen. Al respecto, los autores consideran que estos debían ser tomados en consideración como parte de las políticas gubernamentales a priorizar en el ámbito sanitario.
La letalidad absoluta reportada por estudiosos del tema10 evidencia la incompatibilidad de esta enfermedad con la vida, la cual en ocasiones ocurre de forma intrauterina y en otras alcanza un promedio de sobrevivencia de tres horas. En este caso, el neonato falleció a los 40 minutos de haber nacido.
CONCLUSIONES
El caso que se presenta se enmarca en los estándares teóricos alrededor del Síndrome de Meckel Gruber; en este, estuvieron presentes los tres criterios de la triada diagnóstica. La ausencia de controles prenatales impidió la detección temprana de la enfermedad, poniendo en riesgo la salud de la madre y evidenciando la necesidad de implementar políticas sanitarias que tributen a la disminución de la mortalidad infantil y morbimortalidad materna por esta causa.
Conflicto de intereses
No existen conflictos de intereses
Responsabilidades éticas
Los autores declaran la no presencia de malevolencia o maleficencia, que en la investigación no se realizó experimentos en seres humanos ni en animales, se respetó la confidencialidad de los datos, además de seguir los protocolos sobre la publicación de datos de pacientes.
REFERENCIAS BIBLIOGRÁFICAS
1. Hernández-Viel V, Gómez-Pérez H, Chavarría-Estenoz D. Síndrome de Meckel - Gruber recurrente. MEDISAN [Internet]. 2016[citado 2017 Jun 14];20(4):504-8. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1029-30192016000400011&lng=es
2. Medina ML, Saldarriaga W, Isaza C, Pachajoa H. Meckel syndrome with omphalocele and cleft lip. Rev Cubana Obstet Ginecol [Internet]. 2014[citado 2017 Jun 14];40(2):272-8. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0138-600X2014000200014&lng=es
3. Martínez-Medel J, Sanz-Asín O, Amat-Villegas I, Azcona-Ruiz B, Cabistany-Esqué AC, Martín-de Marcos E. Síndrome de Meckel. Diagnóstico prenatal y diagnóstico diferencial. Prog de Obstet y Ginec. 2012;55(6):269-73.
4. Medina-María L, Saldarriaga W, Isaza C, Pachajoa H. Meckel syndrome with omphalocele and cleft lip. Rev Cubana Obstet Ginecol [Internet]. 2014[citado 2017 Jun 13];40(2):272-8. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0138-600X2014000200014&lng=es
5. Audifred-Salomón J, Barrita-Domínguez IJ, Ortiz de Zárate-Alarcón G, Sánchez-Hernández H, Camacho-Cervantes A. Diagnóstico prenatal de síndrome de Meckel-Gruber. Reporte de un caso y revisión de la bibliografía. Ginecol Obstet Mex. 2016;84(2):105-11.
6. García-Morales AM, Juárez-Azpilcueta A, Durán-Padilla MA, Islas-Domínguez LP, Valdés-Miranda JM. Síndrome de Meckel-Gruber: reporte de un caso de autopsia. Rev Mex Pediatr. 2005;72(5):240-2.
7. Mohamed S, Ibrahim F, Kamil K, Satti SA. Meckel-Gruber syndrome: Antenatal diagnosis and ethical perspectives. Sudan J Paediatr. 2012;12:70-2.
8. Martínez-Medel J, Sanz-Asín O, Amat-Villegas I, Azcona-Ruiz B, Cabistany-Esqué AC, De Marcos EM. Síndrome de Meckel. Diagnóstico prenatal y diagnóstico diferencial. Prog Obstet Ginecol. 2012;55(6):269-73.
9. Prasad U, Prasad U, Sushma J, Rema N. Meckel-Gruber Syndrome fatal disorder - A rare case report with review of literature. International Journal of Dental and Medical Specialty. 2015;2(1):14-7.
10. Aslan K, Aslan EK, Orhan A, Atalay MA. Meckel Gruber syndrome, A case report. Organogenesis [Internet]. 2015[citado 2017 Jun 14];11(2):87-92. Disponible en: http://www.tandfonline.com/doi/full/10.1080/15476278.2015.1055431
11. Sánchez MM, Tejerizo A, Teijelo AI, García RM, Leiva A, García MA, Pérez-Escanilla JA, Benavente JM, Corredera F, Tejerizo-López LC. Síndrome de Meckel-Gruber. Clin Invest Gin Obst [Internet]. 2001[citado 2017 Jun 14];28(7):280-9. Disponible en: http://www.sciencedirect.com/science/article/pii/S0210573X01771080
12. Suárez-Obando F, Ordóñez-Vásquez A, Zarante I. Defectos del tubo neural y ácido fólico: patogenia, metabolismo y desarrollo embriológico. Revisión de la literatura. Rev Colombiana Obstet Ginecol [Internet]. 2010[citado 11 jun 2017];61(1):49-60. Disponible en: http://www.scielo.org.co/scielo.php?script=sci_arttext&pid=S0034-74342010000100007
13. Uysal F, Uysal A. Meckel-Gruber Syndrome with unilateral renal agenesis. J Coll Physicians Surg Pak [Internet]. 2015[citado 2017 Jun 12];25(1):56-7. Disponible: https://www.jcpsp.pk/archive/2015/SS_Apr2015/24.pdf
14. Quiñones-Maza OL, Méndez-Rosado LA, Quintana-Aguilar J, Suárez-Mayedo U, García-Rodríguez M, Barrios-Martínez A, et al. Structural chromosomal rearrangements in prenatal and postnatal cytogenetic diagnosis according to their origin. Rev Cubana Obstet Ginecol [Internet]. 2015[citado 2017 Jun 14];41(1):3-12. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0138-600X2015000100002&lng=es
Recibido: 17 de agosto de 2017.
Aprobado: 17 de septiembre de 2017.
Yosbanys Roque Herrera. Universidad Nacional de Chimborazo, Riobamba. Chimborazo, Ecuador.
Correo electrónico: yroque@hotmail.com

