










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Pediatría
On-line version ISSN 1561-3119
Rev Cubana Pediatr vol.82 no.3 Ciudad de la Habana July-Sept. 2010
Hipoacusia neurosensorial en un síndrome de Noonan y secuencia Poland
Neurosensory hypoacusis in a Noonan's syndrome and Poland's sequence
Julianis Loraine Quintero Noa,I Débora Aleida García Martínez,II María del Carmen Hernández Cordero,III Lidia Báez Allende,IV Orlando Valls Pérez V
IEspecialista de II Grado en Otorrinolaringología. Máster en Atención Integral al Niño. Profesora Consultante. Profesora Auxiliar. Hospital Pediátrico Docente «William Soler». La Habana, Cuba.
IIEspecialista de II Grado en Neonatología. Máster en Atención Integral al Niño. Profesora Consultante. Profesora Titular de Pediatría. Hospital Pediátrico Docente «William Soler». La Habana, Cuba.
IIIEspecialista en Neurofisiología Clínica. Centro de Neurociencias de Cuba. La Habana, Cuba.
IVEspecialista en Neurofisiología Clínica. Hospital Pediátrico Docente «William Soler». La Habana, Cuba.
VDoctor en Ciencias Médicas. Especialista de II Grado en Radiología. Profesor Titular. Hospital Pediátrico Docente «Hermanos Ameijeiras». La Habana, Cuba.
RESUMEN
Se calcula que el 50 % de los casos de sordera profunda en la infancia puede ser de origen genético. Se presenta el caso de un niño de 9 años, atendido en los Servicios de Otorrinolaringología y Genética del Hospital Pediátrico Docente «William Soler», por presentar hipoacusia neurosensorial grave unilateral y displasia congénita de Mondini en el oído izquierdo, del lado contrario a la hipoplasia del músculo pectoral mayor, lo cual coincide con un síndrome de Noonan y secuencia de Poland, que resulta de especial interés. Se constató la hipoacusia con audiometría tonal y potencial evocado auditivo de tallo cerebral. En la tomografía del oído se observó una hipoplasia coclear con agenesia de la espira apical. Se destacan las manifestaciones clínicas y la importancia del estudio otológico e imaginólogico en el diagnóstico de la pérdida auditiva.
Palabras clave: Hipoacusia neurosensorial grave unilateral, displasia de Mondini, síndrome de Noonan, secuencia de Poland.
ABSTRACT
It is estimated that the 50% of cases of deep deafness during childhood may be or genetic origin. This is the case of a child aged 9 seen in Otorhinolaryngology and Genetics Services of the "Wiliam Soler" Teaching Children Hospital due to a unilateral severe neurosensory hypoacusis and Mondini's congenital dysplasia in left ear contralateral to the major pectoral muscle hypoplasia, an interesting situation. Hypoacusis was confirmed using tone audiometry and auditory evoked potential of brain stem. Ear tomography demonstrated a cochlear hypoplasia with agenesis of apical spiral. The clinical manifestations and the significance of the ontological and imaging study in diagnosis of auditory loss are emphasized.
Key words: Unilateral severe neurosensory hypoacusis, Mondini's dysplasia, Noonan's syndrome, Poland's sequence.
INTRODUCCIÓN
Se calcula que el 50 % de los casos de sordera profunda en los niños pueden clasificarse como de origen genético.1-4 El desarrollo y funcionamiento del oído depende de miles de genes y basta la alteración de un solo gen para que se produzca una mutación. Tal complejidad genética es causa de la gran heterogeneidad de la sordera hereditaria. Las sorderas genéticas rara vez son aisladas o forman parte de síndromes que pueden ser de diferentes causas genéticas, cromosómicas, monogénicas o multifactoriales. Las hipoacusias genéticas asociadas o sindrómicas, al ser síndromes muy polimorfos, se clasifican en conjunto en: sordera y trastornos esqueléticos, sordera y alteraciones visuales y con trastornos tegumentarios.1
Se describe dentro de los síndromes malformativos múltiples (SMM) asociados a hipoacusia neurosensorial (HNS) y el síndrome de Noonan (SN), ya descrito por Kobilinsky en 1883, según cita Smith.5 Se reporta que ocasionalmente en un discreto porcentaje de éstos (15 %) existen anomalías estructurales del oído interno e HNS, con una incidencia del SN de entre 1:1 000 y 1:2 500 nacidos vivos.1-7
Se describe también en el SN la diátesis hemorrágica, por defectos de la coagulación y en las plaquetas. Déficit de factores de la coagulación XI:C, XII:C y VIII:C, enfermedad de Von Willebrand y trombocitopenia se reportan en la tercera parte de los casos aproximadamente, así como leucemia en edades tempranas.5,7
Las características fenotípicas de este síndrome son las siguientes: baja talla (71 % de los casos), desviación u oblicuidad antimongoloide de las hendiduras palpebrales, orejas de implantación baja, pterigiun colli, implantación baja y en tridente del cabello en la nuca (87 %), anomalías torácicas y cardíacas (65 %), en especial estenosis pulmonar; malformaciones de las manos y los pies (70 %) y retraso mental.1-4 El patrón hereditario dominante autosómico es el más aceptado en este síndrome.1
La secuencia de Poland, descrita por Poland8 en 1841, es un SMM poco frecuente, con afectación muscular, que se caracteriza por hipoplasia o agenesia unilateral del músculo pectoral mayor, del pezón y la aréola del mismo lado, con defecto de la mano ipsolateral o sin él, y no se acompaña de sordera.2,8-10
Se han descrito pacientes con características concomitantes de síndrome de Noonan y de neurofibromatosis (NF) y otros en los que coexisten síntomas de síndrome de Noonan y de síndrome de Leopard, pero no asociado a secuencia de Poland.1,4
Se presenta un caso de especial interés, el de un paciente que fue remitido al Servicio de Otorrinolaringología por presentar hipoacusia y en el que coinciden ambos síndromes, una coincidencia no descrita antes en Cuba.
PRESENTACIÓN DEL CASO
Se trata de un paciente del sexo masculino, color de la piel negra, de 9 años de edad, que acude a la consulta de otorrinolaringología y genética, y se constató al examen físico las características fenotípicas siguientes (figura 1):
• Talla: baja.
• Cara: mala oclusión dentaria, ligera desviación antimongoloide de los ojos.
• Orejas: de implantación baja, más pequeña la derecha.
• Cuello: corto, tortícolis congénita derecha, pterigium colli, implantación baja del cabello en la nuca.
• Tórax: hipoplasia unilateral derecha del músculo pectoral mayor en su haz esternal, mamila derecha hipoplásica, escápula derecha más pequeña y escoliosis dorsal ligera.
• Manos: hipoplasia del pulgar y de la región tenar de la mano derecha.
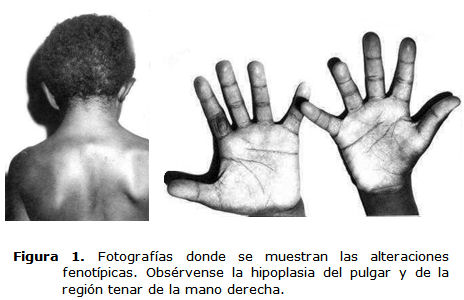
Exploración otorrinolaringológica: Se observó paladar ojival y al examen otomicroscópico, los conductos auditivos externos (CAE) estrechos en ambos oídos y la membrana timpánica (MT) retraída y opaca.
Investigaciones realizadas:
• Ecocardiograma normal.
• Ultrasonografía abdominal normal.
• Estudio radiológico: exploración ósea: anomalías de columna vertebral dorsal, vértebras en mariposa, asimetría de algunos cuerpos vertebrales, sinostosis del 5to. y 6to. arco costal posterior derecho. Aumento de la transparencia del campo pulmonar derecho. Agrandamiento de los cóndilos femorales internos; acortamiento del húmero derecho.
• Cromatina sexual: 0 % cuerpos de Barr. Cariotipo: 46, XY.
Exámenes audiológicos:
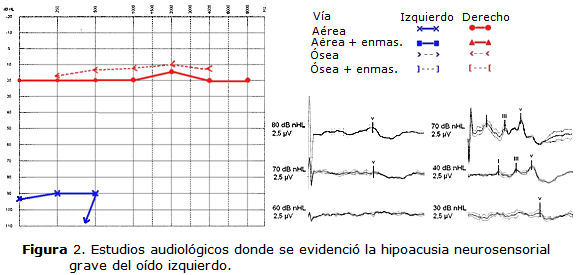
• Audiometría tonal: hipoacusia neurosensorial grave unilateral izquierda. Oído derecho normal.
• Potencial evocado auditivo de tallo cerebral (PEATC):
- Oído derecho: respuesta con morfología típica, latencias absolutas e intervalos interpicos dentro de límites normales, umbral electrofisiológico en 30 dB NHL.
- Oído izquierdo: morfología atípica de los componentes principales I, III y V con umbral electrofisiológico a 70 dB NHL, intervalos interpicos prolongados, compatible con hipoacusia unilateral grave de posible origen neurosensorial y topográficamente con una ubicación coclear de la lesión (figura 2).
Estudio por imágenes:
• En la tomografía del oído se observó hipoplasia coclear con agenesia de la espira apical en el oído izquierdo, y se estableció el diagnóstico de displasia de Mondini. El resto de las estructuras del oído interno se encontraban normales.
• El estudio radiológico del oído derecho fue normal.
DISCUSIÓN
En el síndrome de Noonan se describe la sordera neurosensorial como anomalía ocasional 1-4 y aunque casi siempre las sorderas hereditarias son bilaterales y preferentemente dominante autosómica, también la sordera unilateral puede ser heredada como rasgo autosómico dominante, como se presentó en este caso, o tener otro modo de herencia. Se reporta que el PTPN11 fue el primer gen identificado para el SN y éste se encuentra localizado en el cromosoma 12(12q22qter).3,4 Otras mutaciones en el mismo gen causan el síndrome de Leopard, cuyo cuadro clínico se puede superponer al de Noonan pero en éste además se encuentran lentigenes múltiples acompañando a la sordera.4 En ambos síndromes pueden existir defectos cardíacos tales como estenosis valvular pulmonar y miocardiopatía hipertrófica.4,7 Recientemente han sido identificados 3 genes adicionales en el SN relacionados con el PTPN11.4, 11
En este paciente se constató una hipoacusia unilateral izquierda, del lado contrario a la anomalía del pectoral, la mamila, la tortícolis congénita y la fusión de 5-6 arcos costales, que corresponden con la secuencia de Poland. Cruz y colaboradores describen un predominio en la deformidad de la pared torácica del lado derecho en el 70 % de los casos,2 lo cual coincide en este paciente. La sordera puede aparecer en el SN pero no en la secuencia de Poland, excepto si este último se asocia a síndrome de Möbius.1-5 La cromatina sexual y el cariotipo normal hacen el diagnóstico diferencial con el síndrome de Turner.
Aunque en este caso el ecocardiograma fue normal, se reporta que se pueden asociar a anomalías cardiovasculares tanto el síndrome de Noonan como la secuencia de Poland, y las anomalías cardiovasculares más frecuentes son la estenosis pulmonar en el primero y la dextrocardia aislada en el segundo.3,4
En la timpanometría e impedandiometría se constató una curva timpanométrica patológica con presiones negativas en el oído medio y reflejo estapedial ausente, cuya causa era una obstrucción tubaria grave bilateral. La otitis media asociada a este síndrome se reporta en un 15 %-40 %.7 En la literatura médica revisada no se reporta la hipoacusia en el SN como un hallazgo frecuente y se caracteriza como una HNS de grado variable, bilateral simétrica o unilateral,1,3,4 lo cual coincide con el tipo de sordera del paciente, comprobada por audiometría tonal y PEATC. La pérdida auditiva unilateral izquierda se correlacionó con los hallazgos del estudio tomográfico del oído, donde se constató un desarrollo coclear incompleto con agenesia de la espira apical, por lo que se estableció el diagnóstico de malformación de Mondini del oído izquierdo y su relación con el SN, pues la secuencia Poland es derecha y la asociación Poland-Noonan tiene génesis diferente (Poland-disruptivo y Noonan-monogénico).3,6,8,10
Se reporta que la displasia de Mondini es una malformación congénita del oído interno que usualmente ocurre esporádicamente. Este término ha sido utilizado ampliamente para incluir diversas anormalidades como cóclea plana, conducto coclear pequeño, modiolo hipoplásico, inmadurez en las terminaciones vestibulares y auditivas en órganos o nervios, vestíbulo largo así como canales semicirculares largos, cortos o perdidos. Puede existir aislada o con asociación a otras malformaciones del oído medio u otros órganos. Pueden estar afectados uno o ambos oídos con un rango funcional variable tanto para la porción vestibular como la función auditiva, que es unilateral o bilateral y causada por alteraciones del desarrollo de la cápsula ótica antes de la octava semana de gestación, lo cual resulta como una falta de formación de las porciones más apicales de la cóclea y otras malformaciones en el vestíbulo y los conductos semicirculares debida a una embriopatía, teratogénesis química (talidomida) o infecciosa (rubéola, citomegalovirus), a causa genética, aislada o asociada a un síndrome,12-16 En la literatura revisada se reporta su asociación con los síndromes de DiGeorge, Klippel-Feil, Wildervanck, Pendred, Johanson-Blizzard, Charge, Turner, LAMM (aplasia laberíntica, microtía, microdontía), con trisomías como la 13, 18 o 21 y la displasia de Mondini no sindrómica de tipo familiar.12-15 La evaluación clínica, otológica e imaginológica constituye una herramienta fundamental para el diagnóstico precoz, la caracterización clínica del déficit auditivo y su tratamiento adecuado.
En resumen, este es un caso esporádico ya que no hay ningún otro familiar afectado. Nos proponemos en un futuro continuar profundizando su estudio por su especial interés, al coincidir la hipoacusia y la displasia de Mondini con una asociación sindrómica no descrita previamente en la literatura médica revisada.
REFERENCIAS BIBLIOGRÁFICAS
1. Gil-Carcedo Sañudo E, Vallejo Valdezate LA, Gil Carcedo LM, Menéndez Argüelles ME. Clasificación de las hipoacusias. Hipoacusia neurosensorial. En: Vallejo Valdezate L, Vallejo Valdezate L, Gil Carcedo García LM. Hipoacusia neurosensorial. España: Elsevier; 2003. Pp. 21-32.
2. Cruz-Hernández M, Pastor X, Ballesta F. Dismorfología. Síndromes malformativos múltiples. En: Cruz M. Tratado de Pediatría. T1. España: Océano; 2007. Pp. 258-68.
3. Neal Sondheimer MD. Síndrome de Noonan. [Monografía en Internet]. Disponible en: http://www.healthbasis.com/spanish%20health%20illustrated %20encyclopedia /5/ 001656.htm [citado: 8 septiembre 2009].
4. Formigari R, Michielon G, Digilio MC, Piacentini G, Carotti A, Giardini A, et al. Genetic syndromes and congenital heart defects: how is surgical management affected? Eur J Cardiothorac Surg 2009;35:606-14.
5. Jones KL. Moderate Short Stature, Facial, ± Genital. En: Smith's Recognizable Patterns of Human Malformation. USA: Elsevier; 2006. Pp. 124-7.
6. Allanson JE. Noonan syndrome. Am J Med Genet C Sem Med Genet 2007;145C(3):274-9.
7. Van der Burgt I. Noonan syndrome. Orphanet J Rare Dis 2007;2:4-6.
8. Poland A. Deficiency of the pectoral muscles. Guy's Hosp Rep 1841;6:191-2.
9. Borbolla L, Vázquez B, Lantigua A. Síndrome de Poland. Estudio dermatoglífico de 12 pacientes. Rev Cubana Pediatr 1979;51:15-23.
10. Elli M, Ogmr G, Dagdemir A, Pinarli G, Ceyhan M Daðcinar A. Poland syndrome with intracraneal germ cell tumor in a child. Pediatr Hematol Oncol 2009;26:150-6.
11. Krysiak R, Gdula-Dymek A, Bednarska-Czerwinska A, Okopien B. Growth hormone therapy in children and adults. Pharmacol Rep 2007;59:500-16.
12. Balcero C, Gutiérrez OA. Displasia de Mondini. A propósito de un caso. Acta Otorrinolaringol Cir Cabeza Cuello 2000;28(1):67-70.
13. Glueckert R, Rask-Andersen H, Sergi C, Schmutzhard J, Mueller B, Beckmann F, et al. Histology and synchrotron radiation-based microtomography of the inner ear in a molecularly confirmed case of Charge syndrome. Am J Med Genet A. 2010;152A(3):665-73.
14. Fish JH, Schwentner I, Schmutzhard J, Abraham I, Ciorba A, Martini A, et al. Morphology studies of the human fetal cochlea in Turner syndrome. Ear Hear 2009; 30(1):143-6.
15. Ramsebner R, Ludwig M, Parzefall T, Lucas T, Baumgartner WD, Bodamer O, et al. A FGF3 mutation associated with differential inner ear malformation, microtia, and microdontia. Laryngoscope 2010;120(2):359-64.
16. Griffith AJ, Telian SA, Downs C, Gorski JL, Gebarski SS, Lalwani AK, Sheldon S. Familial Mondini Dysplasia. Laryngoscope 1998 Sept;108(9):1368-73.
Recibido: 2 de mayo de 2010.
Aprobado: 16 de agosto de 2010.
Julianis Loraine Quintero Noa. Hospital Pediátrico Docente «William Soler». Calle 100 y Perla. Altahabana, Boyeros. La Habana, Cuba. CP 10800.
Correo electrónico: julianis@infomed.sld.cu

