










 Custom services
Custom services Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
En la historia se recoge desde el año 1563, la anatomía de la glándula suprarrenal, descrita por Bartolomeo Eustaquio, pero hasta que Thomas Addison en 1855, citado por Stewart y Newell Price.1) realizara su investigación pionera y describiera sus hallazgos clínicos y necroscópicos de 11 casos de enfermedad de Addison, no se definieron con exactitud las funciones de las glándulas suprarrenales.
Un año después, Brown-Sequard, citado por Stewart y Newell Price,1) después de realizar suprarrenalectomía en animales, demostró que las glándulas suprarrenales eran órganos esenciales para la vida.
Las glándulas suprarrenales se dividen en corteza y medula. En la corteza se secretan tres clases de hormonas diferentes: los glucocorticoides, mineralocorticoides y andrógenos; en la médula se producen las catecolaminas. Todas ellas intervienen en múltiples funciones corporales. Se ha estimado que el nivel de secreción al día de cortisol oscila entre 5,7 y 8,7 mg/ml. En cambio, en situaciones de estrés, la tasa de secreción debe ascender a más del doble o triple de lo normal.1,2
Dentro de las enfermedades que pueden afectar la corteza adrenal se describen la insuficiencia suprarrenal primaria, producida por diversas causas como: hiperplasia adrenal congénita, adrenalitis autoinmune (enfermedad de Addison) y uso excesivo de esteroides. El síndrome de Cushing y otros tumores funcionantes o no, pueden ser afecciones que también se pueden encontrar.1,2
El objetivo de este trabajo es mostrar diferentes formas de presentación de algunas de las enfermedades de la corteza adrenal.
Presentación de casos
Presentamos tres casos clínicos que fueron atendidos en el Hospital Pediátrico Universitario “William Soler Ledea”, en diferentes momentos y con cuadros clínicos diversos.
Caso 1
Paciente de 7 años masculino, con antecedentes de hipotiroidismo en tratamiento desde los 4 años. Se ingresa por presentar decaimiento, inapetencia y escaso crecimiento pondoestatural. Se le realizaron estudios de hemograma completo, química sanguínea compuesta por: glucemia, colesterol, triglicéridos, proteínas totales, albúmina, creatinina y enzimas hepáticas, además de estudios hormonales, donde se detectó alteración en las concentraciones basales de cortisol. Se diagnostica insuficiencia adrenal primaria.
Se muestran los resultados de los análisis realizados:
Estudios de química:
Glicemia: 3,6 mmol/L
Colesterol: 4,4 mmol/L
Triglicéridos: 1,0 mmol/L
Proteínas totales: 79 g/L
Albumina: 57 g/L
Creatinina: 39.4 mmol/L
Alanina aminotrasferasa: 16 UI/L
Aspartato aminotransferasa: 30 UI/L
Fosfatasa alcalina: 570 UI/L
Gamma glutamil transpeptidasa: 25 UI/L
Hemograma:
Hemoglobina: 152 g/L
Leucocitos: 3,7 %
Eritrosedimentación: 14 mm/hora
Estudios hormonales:
TSH (hormona estimulante del tiroides): 1,6 nmol/L
Ac. Anti-TPO (anticuerpos antitiroideos): 247 mU/L
ACTH (hormona adrenocorticotropa): 154 pg/ml
Cortisol (8 am): 86 nmol/L
Dos años más tarde, a los 9 años, reingresa por escaso crecimiento a pesar de realizar el tratamiento de manera estricta para la insuficiencia adrenal y mantenerse con dosis adecuadas de hidrocortisona. Se le realizan nuevos estudios, cuyos resultados informan química sanguínea normal y algunas alteraciones en los complementarios de imagen y los hormonales. Se muestran estos datos.
Estudios de imagen:
Radiografía de edad ósea: 2 años y 8 meses.
Tomografía axial computarizada de cráneo: no se observan alteraciones sellares
Estudios hormonales:
Se diagnosticó, en esta ocasión, déficit de hormona del crecimiento, por los resultados de los estudios realizados asociados al cuadro clínico de escaso crecimiento. En este paciente se planteó la posibilidad de un síndrome pluriglandular autoinmune tipo II, por la asociación de 3 déficit hormonales, la deficiencia de cortisol, de hormona tiroidea y de crecimiento.
Caso 2
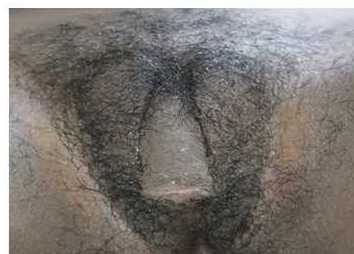
Paciente femenina de 8 años y un mes de edad, con antecedentes de hiperplasia adrenal congénita por déficit de 21 hidroxilasa, forma virilizante simple. Al interrogatorio se recoge historia de cuadros de vómitos y deshidratación asociados a procesos infecciosos, de manera esporádica. Al examen físico presenta aceleración de la talla, mamas en estadio de Tanner 1 (sin progreso) y desarrollo del vello pubiano en estadio de Tanner 4 (vello rizado, abundante, pigmentado, que abarca el pubis en forma de triángulo invertido, sin llegar a la cara interna de los muslos), asociado a aumento del clítoris de manera exagerada. No se le administró el tratamiento indicado con hidrocortisona, durante 5 años. Se le realizaron estudios generales y específicos y se encontró alteración de la hormona 17 hidroxiprogesterona, más de 3 veces su valor normal. La niña mostró desarrollo sexual precoz con virilización de los genitales externos, secundaria a hiperplasia adrenal congénita descontrolada por insuficiencia en el tratamiento. La fotografía fue tomada en acuerdo con los padres de la menor (Fig. 1).
Se muestran los datos.
Estudios de química:
Estudios hormonales:
17 hidroxiprogesterona: 189 ng/dl
Cortisol: 252 nmol/L
TSH: 1.36 mUI/ml
FSH (hormona folículo estimulante):1,0 mUI/ml
LH (hormona luteinizante):1,3 mUI/ml
Caso 3
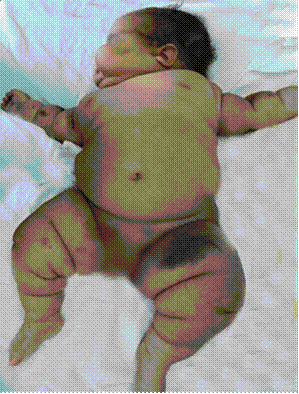
Lactante de 5 meses, femenina, de la raza blanca, sin antecedentes de enfermedad hasta su evaluación. Hija de madre adolescente de 18 años, con antecedentes de salud. Producto de parto eutócico a las 40,1 semanas de gestación, con peso de 3400 gramos. Alimentación con lactancia materna exclusiva. Es atendida por obesidad importante de aparición brusca, en los últimos 3 meses. En el examen físico se observa cara de luna llena y obesidad centrípeta; se constata peso de 9000 gramos, talla: 57 cm, circunferencia cefálica: 44 cm. Relación peso/talla: mayor de 97 percentil; relación talla/edad: menor del 3 percentil; relación peso/edad: 97 percentil. En la piel ligero eritema facial y livedo reticularis. Aparato respiratorio sin alteraciones. Aparato cardiovascular con ruidos rítmicos, de buen tono, pulso fuerte, llene capilar adecuado menor de 3 segundos, tensión arterial: 110/70. Frecuencia cardiaca: 130 latidos por minuto. Abdomen: globuloso, no se define visceromegalia. Sistema nervioso sin alteraciones. Adrenales: no existe vello axilar ni pubiano, estadio de Tanner 1.
Se realizaron investigaciones. Se muestran los datos.
Estudios complementarios:
Hemoglobina: 136 g/l
Glucemia: 4,6 mmol/L
Alanina aminotransferasa: 75 UI/L
Aspartato aminotransferasa: 46 UI/L
Lactato deshidrogenasa: 1615 UI/L
Estudios específicos:
Cortisol en ayunas en 892 nmol/L
Cortisol 11 pm: 920 nmol/L
Sodio: 115.8 mmol/L
Potasio: 4.22 mmol/L
Ecografía de abdomen: hígado homogéneo, vesícula no visualizada, vías biliares no dilatadas, páncreas y bazo normal, riñón izquierdo sin alteración, riñón derecho desplazado hacia abajo, de tamaño normal, parénquima sin alteración, en polo superior en proyección de la glándula suprarrenal derecha se observa masa compleja, predominantemente ecogénica que mide 52 × 42 mm, con zonas de necrosis y calcificación en su interior.
Tomografía axial computarizada de abdomen contrastado: hígado homogéneo que rebasa 4,5 cm el reborde costal, vesícula de paredes finas sin litiasis, vías biliares sin alteración. Bazo y páncreas de aspecto tomográfico normal. Riñón izquierdo de tamaño y estructura normal, riñón derecho de tamaño normal, desplazado hacia la línea media y abajo, con buena diferenciación seno parénquima, sin pielocalectasia. Lesión tumoral en proyección de la glándula suprarrenal derecha hiperdensa, heterogénea, densidades entre 21 y 53 Unidades Hounsfield (UH), con pequeñas áreas hipodensas (1-19 UH), que mide 65 × 44 × 41 mm contornos regulares, bien definidos, desplaza al riñón derecho, sin producir infiltración de dicha lesión. Se diagnosticó tumor suprarrenal productor de cortisol (síndrome de Cushing) (Fig. 2).
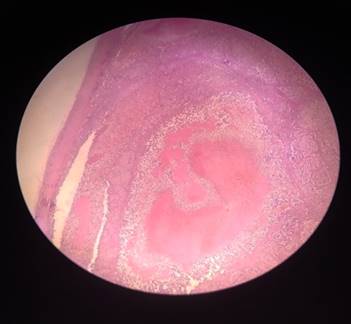
La paciente fue operada por grupo de cirujanos expertos por técnica mínimamente invasiva y se extrajo toda la lesión. La biopsia informó carcinoma adrenal derecho (Fig. 3).
Discusión
Las enfermedades adrenales son infrecuentes de manera general, pero diferentes entidades pueden afectar a las mismas, dando cuadros clínicos diversos.1,2
La insuficiencia suprarrenal primaria, es la incapacidad de las adrenales para secretar una cantidad adecuada de cortisol, fue descrita por primera vez por Thomas Addison a quien debe su nombre. Es una enfermedad con incidencia baja a nivel mundial; menos de 1 por 100 000 habitantes, con una prevalencia de 4-6 por cada 100 000 personas y afecta a ambos sexos por igual.1,2,3
La etiología autoinmune es la más frecuente (adrenalitis autoinmune) y puede darse de forma aislada o como parte de un síndrome pluriglandular autoinmune tipo 1 o tipo 2. La mitad de los pacientes con insuficiencia adrenal tienen al menos otra glándula endocrina comprometida, por lo cual se deben realizar estudios de pesquisa para detectar otras alteraciones endocrinas en estos pacientes.2,4,5
A pesar de su baja incidencia, vemos en el primer caso clínico presentado que el motivo de consulta fue el escaso crecimiento pondoestatural, causa de control frecuente en pediatría. En este paciente se asociaron además otros déficit hormonales; tirotropa y de crecimiento, por lo cual se debe tener en cuenta la posibilidad de estar en presencia de un síndrome pluriglandular autoinmune tipo II (SPA II).
Los SPA son enfermedades raras que se caracterizan por la asociación de al menos dos déficit hormonales por insuficiencia glandular, mediadas por mecanismos autoinmunes.2
En el caso previamente presentado no hubo posibilidad de dosificar todos los anticuerpos específicos, como anticuerpos antiadrenal para confirmar el diagnóstico de adrenalitis autoinmune. En el paciente si se detectó elevación de los anticuerpos antiperoxidasa tiroidea (Ac. Anti-TPO), hecho que nos indica la presencia de alteración inmunitaria.
El segundo paciente, se trata de una niña con diagnóstico temprano de hiperplasia adrenal congénita (HAC) por déficit de 21 hidroxilasa.
La HAC tiene 5 variantes descritas, según el déficit enzimático que se presente. La forma más común es por deficiencia de la enzima 21 hidroxilasa, necesaria para la síntesis de cortisol y aldosterona.6,7,8,9. La enfermedad tiene una presentación mundial que oscila entre 1: 10 000 a 1: 20 000, con una incidencia anual de estimada entre 1:5 000 a 1:15 000.8,9,10
A pesar de identificar el problema de manera adecuada e indicar el tratamiento oportunamente, el actuar negligente de los familiares, conduce a una complicación mayor: la virilización de los genitales asociado a pubertad precoz.
En la paciente con síndrome de Cushing por carcinoma adrenal secretor de cortisol, lo llamativo es la edad de presentación de este tipo de lesión, muy infrecuente en la etapa de lactante.
La literatura describe una incidencia de 0,5 a 2 casos por millón de habitantes al año.11,12,13,14 Los datos del Nacional Cancer Institute de EE. UU. estiman que solamente 1,3 % de los procesos de la infancia son carcinomas.11
Se describe que las lesiones malignas de las suprarrenales tienen una evolución muy agresiva y 79,0 % son funcionantes, pueden secretar sexo esteroides y provocar virilización o feminización dependiendo de si afecta a mujeres u hombres, respectivamente. El síndrome de Cushing es una de las formas habituales de presentación.14,15,16,17
La niña fue operada y aunque luego siguió control por oncología, se logró la reducción de los niveles de cortisol luego de la extirpación de la tumoración.
Se concluye que es importante tener en cuenta las diferentes formas de presentación de algunas enfermedades que afectan a la corteza suprarrenal que podemos encontrar en la práctica clínica diaria para poder tener claridad a la hora de atender a pacientes con síntomas y signos tan comunes como el decaimiento, escaso crecimiento, vómitos o el aumento de peso brusco.

