










 Custom services
Custom services Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
El factor XII (FXII) o factor de Hageman, es una glicoproteína de cadena simple que circula en el plasma como un zimógeno, su concentración es de 40 ug/ml y se activa al entrar en contacto con superficies aniónicas que provocan un cambio conformacional en presencia de iones de zinc. Su activación provoca un cambio a una molécula de dos cadenas: una pesada de 353 aminoácidos (aa) y una ligera de 243 aa, unidas entre sí por un enlace disulfuro ubicado entre los residuos Cys340 y Cys367. La cadena pesada se encarga de la unión a las superficies aniónicos y la cadena ligera a la actividad proteolítica.1
En el modelo clásico de la cascada de la coagulación el FXII o factor de Hageman es activado tras su exposición a superficies con carga negativa presentes en la membrana basal del endotelio a través de los llamados factores de contacto (calicreína y quininógeno de alto peso molecular) (vía extrínseca). Tras su activación se genera una activación secundaria de los factores XI, IX y X. De forma simultánea tras el daño endotelial, se activa el factor tisular y se forma el complejo factor tisular/factor VII activado, que a su vez genera una activación secundaria del factor X y IX (vía intrínseca). Una vez activados el factor X y el factor IX se pone en marcha la vía común a través de la activación del factor II (trombina), que genera la transformación de fibrinógeno a fibrina responsable de la formación del coágulo y a su vez la activación del factor XIII responsable de la estabilización del coágulo.1
El modelo clásico de la coagulación permite definir los estudios generales de laboratorio, sin embargo, no considera la interacción de la vía extrínseca y la vía intrínseca por lo que no explica mecanismos de la hemostasia in vivo. Por su parte, el modelo celular de la coagulación propone a la vía intrínseca como un amplificador iniciado por la vía extrínseca a través de tres fases: la fase de iniciación desencadenada por el daño endotelial, el cual genera de forma simultanea la formación del complejo factor VII activado/factor tisular y la activación del factor XII tras su exposición a superficies con carga negativa de la membrana basal del endotelio.1
La deficiencia del factor XII es rara y se presenta en uno de un millón de individuos, de herencia autosómica recesiva o adquirida y no suele manifestarse clínicamente puesto que no produce trastornos hemorrágicos como ocurre en la deficiencia de otros factores.2
EL objetivo de esta presentación es describir las manifestaciones clínicas de un lactante con diagnóstico de deficiencia de factor XII de la coagulación.
Presentación del caso
Se trata de un paciente de 10 meses, nacido a término, vía vaginal, con adecuada antropometría, sin antecedentes patológicos o familiares de importancia. Ingresa en la consulta por aparición de equimosis espontánea de dos días de evolución, inicialmente en muslos con mejoría parcial y posteriormente en región periumbilical, la cual persistió y no estaba asociada a trauma ni a otra sintomatología, con un tiempo de tromboplastina parcial activado (TPTa) prolongado y un FXII deficiente. En la valoración inicial se comprobó equimosis periumbilical (Fig. 1) y área de hiperpigmentación en cara interna del muslo izquierdo, sin adenopatías ni otros hallazgos anormales. Por lo anterior, se planteó como impresión diagnóstica discrasia sanguínea a descartar; se solicitó hemograma extendido de sangre periférica y ferritina.
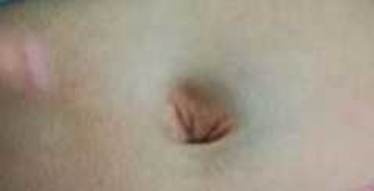
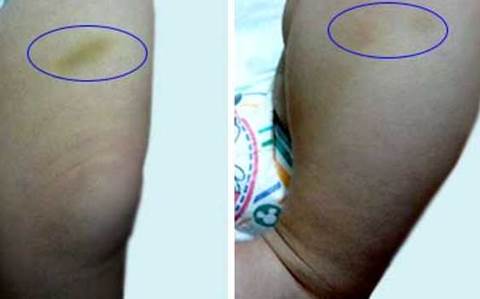
Después de un mes se hizo control al paciente en consulta, con persistencia de equimosis a nivel de abdomen y miembros inferiores (Fig. 2), las cuales cedían de forma espontánea y no se observaban durante la exploración física.
Se revisaron los resultados paraclínicos: hemograma con anemia microcítica hipocrómica leve sin alteraciones en línea granulopoyética ni megacariopoyética, extendido de sangre periférica con hipocromía ligera, anisocitosis y microcitosis, y concentraciones bajas de ferritina. Se hizo diagnóstico de anemia ferropénica, se inició tratamiento con suplemento de hierro y se amplió el estudio con determinaciones de: tiempo de protrombina de 11,2 s (control de 11,3 s), TPTa de 39,5 s (control de 28,5 s), coombs directo negativo y ecografía abdominal, resultados normales en todas las pruebas, asímismo se solicitó dosificación de factores con los siguientes resultados: factor IX: 82,6 %, factor XI: 97,5 %, factor XII: de 41 % y TPTa: 36,5 s (control de 30 s). Se hizo el diagnóstico de deficiencia de FXII. Por lo anterior, continuó en seguimiento por consulta externa de pediatría y no se registraron otras manifestaciones clínicas.
Discusión
Este caso se presenta porque la deficiencia del FXII es un desorden poco común que no está asociado con manifestaciones hemorrágicas, por lo que en un lactante menor se deben considerar otras causas más frecuentes como trauma y sepsis. En este caso no hubo hallazgos clínicos adicionales como deformidad o dolor en reja costal o en extremidades que hiciesen pensar en trauma y tampoco fiebre u otros signos de respuesta inflamatoria sistémica que hiciesen pensar en sepsis.
Se realizó un enfoque general del síndrome hemorragiparo sin demostrar alteraciones en el número ni en la morfología plaquetaria, que pudiesen sugerir una alteración en la hemostasia primaria, y un TPTa prolongado con un TP normal sugerente de un trastorno en la hemostasia secundaria, específicamente en la vía intrínseca de la coagulación (factores VIII, IX, X, XI, XII y factores de contacto). Si bien no se realizó dosificación de factores VIII y X, se documentó una disminución en la actividad del factor XII consistente con prolongación de TPTa. No se estudió a los padres para descartar una deficiencia de tipo congénito (herencia autosómica recesiva) y no hubo hallazgos en la historia clínica sugestivos de deficiencia de tipo adquirido como administración de heparinas o presencia de factores inhibidores, muy frecuentes en casos de infección.
La deficiencia del FXII se describió por primera vez en el año de 1955 por el Dr. Ratnoff, citado por Girolami y otros2) en la caracterización de tres pacientes sin antecedentes personales de importancia, que dentro de estudios prequirúrgicos presentaron un tiempo de coagulación prolongado sin síntomas hemorrágicos. Aproximadamente quince años después, uno de los pacientes caracterizados previamente, ingresó con una fractura con requerimiento de atención quirúrgica, por lo que se documentó nuevamente un tiempo de coagulación prolongado sin que esto implicara complicaciones en el posoperatorio inmediato, sin embargo, hacia el décimo día de hospitalización, el paciente presentó un evento tromboembólico pulmonar y falleció. Por lo anterior, inicialmente se confirió a esta deficiencia un riesgo trombótico elevado, lo que es posteriormente debatido, teniendo cuenta otros factores de riesgo propios del paciente, tales como fractura de cadera y reposo prolongado.2
Desde el año 1983, Goodnough y otros, publicaron una serie de casos de pacientes con rasgo Hageman (1 % de actividad del FXII) que tuvieron antecedente de trombosis, evento coronario agudo y enfermedad de Moyamoya.2) En 1984 Lodi, citado por Govers-Riemslag3) publicaron un caso en el que describen deficiencia severa del FXII (menor del 1 % de actividad) y evento coronario agudo en paciente de 40 años. Por último, en 1991 Lämmle citado por Petousis y otros,4) dieron a conocer un estudio de 74 pacientes suizos, y se sugirió la asociación de la deficiencia homocigota del FXII con un mayor riesgo de trombosis venosa.
Estudios actuales como los que plantea Keene, citado por Bender y otros,1) y Matafonov citado por Zilberman-Rudenko y otros,5) realizados en modelos murinos y babuinos, plantean deficiencia de FXII y riesgo aumentado en la incidencia de eventos trombóticos, sin embargo, estudios clínicos controlados recientes en humanos no han logrado encontrar una asociación entre estos dos hechos.6
A pesar de lo anterior, se cuenta con nuevas descripciones en la literatura como el caso descrito en el año 2019 en el hospital King Salman de Arabia Saudita en el que una paciente de 29 años, a la tercera semana de posoperatorio de cesárea, ingresa en el departamento de urgencias por disnea severa y dolor torácico. Se documenta embolia pulmonar masiva con un tiempo de tromboplastina prolongado y una actividad del FXII deficiente.7
Esta deficiencia suele diagnosticarse en la edad adulta durante estudios prequirúrgicos o estudios de rutina ya que, aunque cursa con TPTa prolongado no se manifiestan síntomas indiscutibles de sangrado como es demostrado en diversos casos.
En el Departamento de Medicina Interna del Hospital Hamman en Catar, se documentó un TPTa prolongado y finalmente una deficiencia de factor de Hageman en un paciente de 37 años admitido por pancreatitis aguda. El paciente había presentado un episodio de gingivorragia el año previo a su ingreso atribuido a periodontitis. No había presentado ningún evento trombótico.8) Así mismo en el departamento de medicina interna de la Universidad de Dakota del Norte durante la toma de laboratorios prequirúrgicos en un paciente de 75 años sin antecedentes de eventos tromboembólicos ni hemorrágicos quien cursaba con una fractura intertrocantérica de fémur izquierdo secundaria a trauma, se documentó un TPTa persistentemente prolongado con actividad del factor de Hageman disminuida.9
A diferencia de lo descrito en el Hospital Hamman en Catar8) y en el deparamento de medicina interna de la Universidad de Dakota del Norte,9 en el presente caso se hace diagnóstico de deficiencia del factor de Hagemann como parte del estudio del síndrome hemorragiparo en un lactante con manifestaciones de sangrado. Allí radica la importancia de reportarlo en la literatura.
Se concluye que a pesar de no ser común, la deficiencia del factor XII puede estar asociada a manifestaciones hemorrágicas como equimosis, tal como lo descrito en el presente caso.

