










 Custom services
Custom services Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
El abordaje de las diarreas en el lactante comprende múltiples entidades relacionadas con mecanismos de maldigestión y malabsorción intestinal.1,2 En este contexto las enteropatías congénitas representan un grupo de afecciones heterogéneas que son raramente diagnosticadas, algunas documentadas en las últimas décadas; con presentación clínica de diarreas intratables, de comienzo habitual en el curso del primer año de edad, aunque generalmente surgen en el período neonatal y los primeros tres meses de vida difíciles para el diagnóstico y atención terapéutica fundamentado en el enfoque de nuevos conceptos y conductas.3,4).
El rango de las referidas enfermedades congénitas varía desde afecciones simples, como la intolerancia a las proteínas de la leche de vaca hasta defectos de polarización de la membrana del intestino de elevada letalidad que pueden hasta requerir por fallo del órgano de un trasplante del intestino.5).Asímismo, los resultados en los avances del conocimiento en genómica humana han aportado comprensión acerca de las correlaciones genotípicas y fenotípicas, lo cual ha repercutido en la cuidado de las afecciones diarreicas congénitas del intestino.1).
El desarrollo del arte de la práctica pediátrica y la gastroenterología pediátrica como subespecialidad, han logrado a la luz de los avances alcanzados, los fundamentos para la salud de la infancia y adolescencia basados en los aportes de las ciencias médicas para establecer con prontitud el diagnóstico y tratamiento de enfermedades poco reconocidas, que permitan evitar una desfavorable evolución de la morbilidad en distintas direcciones, en especial en la repercusión del estado de nutrición.6).
El objetivo de este artículo es examinar los conocimientos más recientes para la orientación diagnóstica de las enteropatías congénitas en el contexto clínico de las diarreas crónicas intratables del período posnatal y primeros meses de vida.
Análisis e integración de la información
Las diarreas crónicas en el niño menor de 6 meses de edad de causa no infecciosa correspondientes a las denominadas enteropatías congénitas son entidades poco reconocidas7).y es en las primeras semanas de vida y en general en el curso de los tres primeros meses de edad, cuando los trastornos congénitos no diagnosticados o raros pueden semejar enfermos en estado crítico con adquiridas afecciones comunes. Ante esta problemática es de importancia la difusión de las características clínicas de dichas entidades para lograr su identificación acertada desde el inicio de la diarrea, en el ámbito de la evaluación de la enfermedad y disminuir el riesgo de un diagnóstico tardío.8).El diagnóstico temprano para obtener una clasificación precisa de la diarrea será decisivo para la salud y la vida del lactante.
Las enfermedades catalogadas como raras se relacionan con su baja prevalencia, afectan un número limitado, muy escaso de personas en comparación con la población general y su propia rareza plantea cuestiones específicas.9,10).En la Unión Europea las definen como todas aquellas cuya cifra de prevalencia se encuentra por debajo de los 5 casos por cada10 000 habitantes, en los EE. UU. se utiliza una cifra global de casos (menos de 200 000 para todo el país). Otras naciones prefieren utilizar una definición más restrictiva como menos de 4 casos por cada 10 000 habitantes.10).
Síndrome de diarrea intratable. Descripción clínica
Es clásica la descripción de Averyy y otros,11sobre el denominado síndrome de diarrea intratable o “diarrea infantil intratable”, descrito como una diarrea crónica severa, prolongada e intratable, a pesar dela extensa terapia hospitalaria. La descripción del síndrome comprende las siguientes características:12
Diarrea de más de dos semanas de duración.
Edad de menos de tres meses.
Tres o más cultivos de heces negativos para patógenos bacterianos.
Lactantes que han recibido hidratación y fluidos endovenosos.
A pesar del tratamiento hospitalario la diarrea es persistente e intratable.
Alta mortalidad.
Se adiciona a los postulados de Avery, el siguiente elemento:
Al momento actual, en la práctica médica el concepto de “diarrea intratable” se ha prolongado a más de tres meses de edad y es usado para la diarrea crónica severa y prolongada asociado a mal nutrición, sin respuesta satisfactoria y difícil atención.13
Para la orientación práctica de la diarrea intratable se reconocen tres formas relacionadas con los eventos de su fisiopatología, correspondientes a diarrea secretora, osmótica y mixta, sin embargo, en años recientes se propone modificar la interpretación de la terminología por imprecisa y sugerido el examen de la composición de las heces como expresión de los mecanismos que determinan el aumento de su producción,5 elemento semiológico vital para orientar la causa de la diarrea crónica (Cuadro 1).
Los conceptos de las tres formas referidas son los siguientes:
Diarrea osmótica
Es la terminología tradicionalmente utilizada para referirse a la diarrea resultante de heces con solutos o nutrientes no absorbidos de la dieta, sin embargo, toda diarrea implica fuerzas osmóticas, por lo tanto, la denominación de “diarrea inducida por la dieta” se ha sugerido por ser más precisa. Se caracteriza por una elevación osmótica en las heces (> 100 mOsm). Lo que puede acontecer en la malabsorción de glucosa o disacáridos (intolerancia a la lactosa y hereditaria a la fructosa) y procesos del colon.
Diarrea secretora
Se ha relacionado con la descripción de la fisiopatología subyacente de las diarreas causadas por la secreción de iones activos en el intestino, pero no contempla a las diarreas acuosas con alto contenido de sal causadas por defectos en la absorción intestinal de sodio, como ocurre en las diarreas congénitas de sodio y también de electrolitos, como la diarrea congénita clorada y en algunas infecciones virales. Tampoco se puede utilizar el término secretor para describir todas las diarreas con una expresión osmótica fecal baja (<50 mOsm) (Tabla 1), porque las heces con efecto osmótico bajo suelen ser el resultado de una combinación de mejora de la secreción de fluidos impulsada por aniones y pérdida de la absorción de fluidos impulsada por sodio. Se ha propuesto utilizar el término “diarrea relacionada con el transporte de electrolitos” según la interpretación de su fisiopatología. Que incluyen enteropatías congénitas de cloruro o sodio.12)
Diarrea mixta
Por último, la diarrea que obviamente no es ni secretora ni osmótica, es frecuente la deposición de las heces con valores intermedios de expresión osmótica (50-100mOsm) y son causadas generalmente por una combinación de los elementos de la diarrea inducida por la dieta y con la relacionada con el transporte de electrolitos, resultante de diferentes ingestas dietéticas.
Cuadro 1 Diferencias entre las diarreas osmótica y secretora
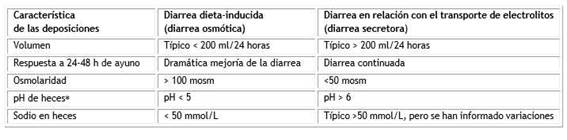
Fórmula osmolaridad= 290 - [2 × (Heces Na() ( (Heces K(); el pH de las heces puede ser variable en ocasiones.
Fuente: Modificado de Elkadri AA. Congenital Diarrheal Syndromes. Clinics in Perinatology 2019.5
Los argumentos relacionados son de trascendencia para la interpretación semiológica de las referidas diarreas osmótica y secretora y permiten orientar por un lado, la causa de la diarrea y por otro, la aplicación en las formas crónicas de un nuevo enfoque basado en las manifestaciones clínicas, respuesta a la supresión de la vía oral y determinación del pH. Estos son procederes rápidos y sin complejidad que ayudan a interpretar la causa de la diarrea junto con la posibilidad de determinar la osmolaridad y presencia de sodio (Na+) en las heces, elementos que pueden proporcionar una mejor evaluación de la naturaleza de la diarrea (Cuadro 1).
Enteropatías congénitas
Las enteropatías congénitas (EC) son trastornos hereditarios monogénicos, con predominio de herencia autosómica recesiva causantes de diarrea crónica grave y persistente que, a menudo conducen a una insuficiencia intestinal con potencial mortalidad. A la luz de los conocimientos descritos se han agrupado en cinco categorías según los criterios de la terminología relacionados con la fisiopatología de las diarreas crónicas.8).
Semiodiagnóstico de las enteropatías congénitas14).
Una serie de indicadores resultan de interés para la orientación de dichas enfermedades, los que se relacionan a continuación:
Historia Familiar de consanguinidad.
Antecedente prenatal de polihidramnio.
Características de las diarreas.
Momento de aparición de la diarrea.
Relación de la diarrea con alimentos/ supresión de la dieta.
Evolución de la diarrea como intratable.
Manifestaciones extradigestivas
Los defectos de las células epiteliales representan el rasgo principal de las enteropatías congénitas e incluye cinco categorías que comparten una fisiopatología común. Se muestra una novedosa clasificación de estas enfermedades propuesta por Elkadri5) (Cuadro 2).
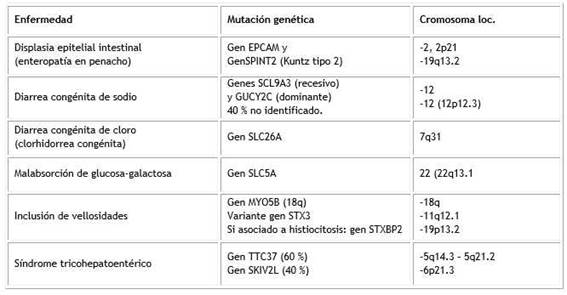
En este contexto sobresalen enfermedades graves, debidas a la categoría de tránsito defectuoso de enterocitos y polaridad, como la inclusión de microvellosidades y la displasia epitelial intestinal que se presenta con diarrea acuosa abundante, desequilibrio electrolíticos y retraso del crecimiento. Las cinco categorías de las EC se han clasificado de la siguiente forma: 5
Clasificación de las diarreas congénitas según etiopatogenia5
1. Defectos en el transporte epitelial de nutrientes y electrolitos
2. Defectos de las enzimas epiteliales y del metabolismo
Deficiencia congénita de lactasa.
Deficiencia congénita de sucrasa-isomaltasa.
Deficiencia de trehalasa.
Deficiencia de enterocinasa.
Deficiencia enzima diacilglicerol-aciltransferasa 1 (DAGT1)
Tamizado de enteropatía perdedora de proteínas.
Abetalipoproteinemia, hipolipoproteinemia, enfermedad de retención de quilomicrones.
Disqueratosis congénita.
Síndrome de Kabuki.
3. Defectos del tránsito epitelial y polaridad
4. Disfunción de las células enteroendocrinas
5. Enteropatía asociada a desregulación
Histopatología intestinal
La interpretación de las biopsias de intestino por parte del patólogo es decisiva para arribar a un diagnóstico certero y con prontitud en las enteropatías infantiles productoras de diarreas intratables de causa congénita. Los hallazgos de las biopsias pueden variar desde una mucosa intestinal normal hasta alteraciones con diferentes grados de atrofia de las vellosidades o inflamación, o características específicas subyacente. Se relacionan anomalías epiteliales, vacuolización de lípidos en los enterocitos, ausencia de células plasmáticas, linfangiectasia, microorganismos e infiltración eosinofílica o de histiocitos de la mucosa.5,13).
Entre las EC que cursan con morfología normal de las vellosidades de la mucosa intestinal, se incluye la diarrea congénita por cloruros, malabsorción de azúcares (glucosa-galactosa) y deficiencia congénita de sacarasa-isomaltasa.
Entre las que cursan con atrofia o inflamación de la lámina propia de las vellosidades, se relacionan la enteropatía autoinmune, la enfermedad de inclusión de microvellosidades, la gastroenteritis eosinofílica, la displasia epitelial intestinal y la enteropatía sensible al gluten, aunque esta última es rara antes del año de edad y requiere la administración de alimentos que contengan gluten por un tiempo mínimo para la aparición de las alteraciones de la mucosa duodeno-yeyunal. Asímismo, entre otros hallazgos o rasgos específicos, se describen los enterocitos llenos de lípidos, ausencia de células plasmáticas (como en la inmunodeficiencia), linfangiectasia (en linfangiectasia intestinal primaria), infiltración de eosinófilos en la lámina propia (en enteritis eosinofílica) y de histiocitos (en histiocitosis).13
Se examina a continuación información esencial de la displasia epitelial intestinal o enteropatía en penacho, de la diarrea congénita de sodio, de la clorhidorrea congénita, malabsorción de glucosa galactosa, enfermedad de inclusión de microvellosidades y síndrome tricohepatoentérico
Displasia epitelial intestinal
La displasia epitelial intestinal (DEI) es una enfermedad genética, de carácter rautosómico recesivo, también denominada enteropatía en penacho correspondiente a la categoría de defecto en el tránsito epitelial y polaridad relacionado con las EC, con manifestaciones clínicas de diarrea intratable, rasgo típico de alteración histopatológica de enterocitos en penacho de la mucosa del duodeno.15).
Esta enfermedad se describió hace apenas algo más de 25 años, en 1994 por Reifen y otros, en Toronto, Canadá,16 en tres niños, con diarrea crónica acuosa severa e intratable, refractaria a la dieta, que requirieron nutrición parenteral total (NPT) con hallazgo típico histológico y ultraestructural de DEI.
En 1995 Goulet y otros,17, en Francia describieron cuatro niños (2 hermanas) con similar hallazgo clínico e histológico de desorganización de la mucosa intestinal apical por penacho. Estos autores, en 1998 ampliaron la descripción de una serie de 47 niños con diarrea intratable, con la presentación de un grupo de 10 niños con lesión similar a lo referido por Reifen,16 algunos de origen árabe y consanguinidad familiar.18),Posteriormente, en 2007 se informó por Davidson la denominada “enteropatía familiar”19),en pacientes de padres consanguíneos y además con vínculo familiar, lo que reafirmó la naturaleza hereditaria de la enfermedad, con descripción similar a la original de 1994.
Epidemiología
Es una enfermedad de distribución mundial, con pocos datos epidemiológicos disponibles. La prevalencia es mayor en las regiones con grado elevado de consanguinidad, con hermanos o primos afectados por la herencia autosómica recesiva, cuyos padres son normales. En 2007, Goulet20 estimó una prevalencia entre 1/ 50 000 a 1/ 100 000 nacimientos en Europa occidental, mientras en la actualidad se informa , como posibilidad, alrededor de 1/ 200 000 nacidos, con mayor incidencia entre la migración árabe con evidente consanguinidad, y predominio en las islas maltesas aunque es más común en los países del Oriente Medio21,22) que en los europeos; recientemente se descubrió en China20).
Se han reconocido en la literatura alrededor de 200 casos.5). Recientemente se informa, en dependencia del tipo y naturaleza de la mutación de la molécula de adhesión epitelial celular (EpCAM, sigla de abreviatura en inglés) 5).que es una glucoproteína transmembrana de adhesión célula-célula homotípica, que se asocia con el síndrome de Lynch.23).
Genética
El carácter genético de la enfermedad está argumentado por la descripción de mutaciones en los genes EpCAM24,25) y serina peptidasa, gen Kunitz tipo 2 [(SPINT2), identificación de la mutación en la molécula de adhesión de células epiteliales(EpCAM) del gen localizado en el cromosoma 2 (2p21)], que codifica la proteína unida a la membrana basolateral de las células epiteliales intestinales, descubierta por Sivagnanam y otros,26 en 2008.
EpCAM es una proteína esencial involucrada en la señalización, diferenciación, migración y proliferación de célula a célula, así como en la adhesión celular independiente del calcio. La mutación es responsable de DEI y también de la forma sindrómica. Años después se identificó la mutación del gen inhibidor de SPINT2, aunque en menor frecuencia. 27). De las mutaciones, el 74 % aproximadamente corresponde a EpCAM y el 28 % a SPINT2. Se han descrito más de 42 mutaciones de EpCAM, que parecen tener un papel vital en la interacción célula-célula mediante la reclutación de α-actinina intracelular y la localización con E-cadherina y claudina-7, en áreas de uniones célula-célula y uniones estrechas, respectivamente.[28,29
Fisiopatología
Las modificaciones por pérdida de expresión del gen EpCAM, por estar mutado o truncado y la proteína disminuida o mal localizada por probable reducción de la estabilidad, asociada con mutación alternativa de SPINT2, representan los indicadores genéticos de la enfermedad.30).
Estudios experimentales en modelos de ratones y humanos realizados en los últimos años han demostrado evidencias de eventos aún no esclarecidos totalmente, en especial en las forma sindrómica y alteraciones de la homeostasis intestinal en el desarrollo de EC. Estos eventos conducen a desorganización estructural de criptas y vellosidades por alteraciones tempranas de las funciones de crecimiento, incluido defectos de la función de barrera, disminución de proteínas de unión estrecha de las células epiteliales y del transporte de electrolitos.
La homeostasis epitelial se mantiene por equilibrio dinámico entre las células epiteliales intestinales y la barrera mucosa, mientras que la diferenciación y composición celular, la barrera y uniones célula-célula, son los elementos principales en la patogenia de EC.5
Síntomas clínicos
La aparición de las manifestaciones es desde los primeros días de nacido,31),con diarrea acuosa profusa (secretora, osmótica o mixta), severa deshidratación y desequilibrio de electrolitos, producida con componentes de diarrea inducida por la dieta y relacionada con el transporte de electrolitos, con malabsorción de nutrientes.5,15).
Los lactantes son irritables y, además de la diarrea intratable, algunos pacientes pueden presentar vómitos, distensión abdominal y15 y pérdida de peso. La repercusión funcional del intestino puede resultar en manifestaciones clínicas de malabsorción, desnutrición, y deterioro del crecimiento (fallo de medro)32) con evolución a fallo intestinal grave, que demanda alimentación parenteral y, en algunos casos se ha informado el trasplante intestinal como solución.4,15).
Forma sindrómica
La asociación de los síntomas intestinales descritos a síntomas extraintestinales, definen la forma sindrómica. La dismorfia facial es rasgo sindrómico, que incluye hipoplasia del tercio medio facial, estrechamiento de las fisuras palpebrales e hipertelorismo (nariz ancha y mandíbula pequeña). La cavidad bucal se describe con paladar arqueado. Asímismo, se han referido subconjuntos de manifestaciones oculares, articulares, óseas, hepáticas, dermatológicas y anomalías (atresias) del intestino.
Entre los síntomas oftalmológicos se ha informado inflamación crónica de la córnea (ojos de color rojo) con trastornos de la visión por desarrollo de queratitis puntuada superficial, cataratas y erosiones corneales.[5,15,18,20].
Entre las atresias, se encuentran las esofágica, anal, intestinal y coanal.[5,15,20),Las óseas corresponden a displasia,15),mientras en el hígado acontece enfermedad colestásica. Entre las manifestaciones extraintestinales, la artritis crónica es más común en los portadores con mutaciones en EPCAM,5,32) mientras las oftalmológicas y la atresia son más frecuentes en pacientes con mutaciones SPINT2.[5]. Los pacientes presentan hipotonía del tronco y de las extremidades inferiores, con baja talla. El desarrollo psicomotor es normal. La ecografía cerebral ha mostrado moderada dilatación ventricular bilateral.15).
Diagnóstico de certeza
Se fundamenta en la combinación de criterios clínicos y hallazgos histopatológicos sobre los cambios estructurales característicos del epitelio de la mucosa del intestino delgado en biopsia del duodeno obtenida por endoscopia gastroduodenal.33). Los hallazgos típicos incluyen atrofia de las vellosidades parcial, subtotal o total e hiperplasia de las criptas, con escasa infiltración de células mononucleares o nula en la lámina propia o linfocitosis intraepitelial.
La anomalía patológica específica consiste en la identificación de los “penachos epiteliales focales”8),producidos por agrupación (apiñamiento) apical de enterocitos desorganizados de las vellosidades de la mucosa intestinal y alteración de la membrana basal. Los enterocitos congregados en la superficie vellositaria (penachos) adoptan forma redondeada, que asemejan una lágrima o papilas pequeñas, que pueden afectar 70 % de las vellosidades. La severidad de la atrofia vellositaria puede ser variable, aunque habitualmente no hay infiltración de células inflamatorias.13). Las microvellosidades intestinales (borde en cepillo) suelen permanecer intactas, a diferencia de la enfermedad de inclusión de vellosidades.
Estudio de inmunotinción para identificación de EpCAM puede ser decisivo cuando los criterios no son evidentes. Asímismo, en ocasiones, en lactantes de edad temprana, la biopsia no es concluyente para identificar la lesión de agrupación focal de los enterocitos (penachos) y, es necesario repetirla en un tiempo posterior para la obtención de otra muestra de mucosa intestinal y poder demostrar el desarrollo de la lesión característica.
Las pruebas moleculares para la demostración de la mutación del gen EpCAM o SPNT2 permiten confirmar la enfermedad33). El diagnóstico genético prenatal puede ser realizado cuando existan antecedente familiar de un primer caso, no obstante, el consejo genético es necesario cuando se ha identificado la enfermedad.
Conducta terapéutica
La nutrición parenteral total (NPT) puede ser imprescindible para la mayoría de los pacientes con DEI, mientras en otros el apoyo parenteral resulta permanente14).El pronóstico a largo plazo puede ser sombrío, en dependencia de la posibilidad de mantener NPT imprescindible. La diarrea intratable y la malabsorción intestinal pueden evolucionar a desnutrición severa y aparición de infecciones asociadas con fallo intestinal irreversible. El procedimiento de trasplante de intestino es la opción terapéutica curativa para obtener la autonomía digestiva con la consiguiente homeostasis intestinal, solución reparadora que es una posibilidad limitada en la actualidad a escasos países de alto desarrollo.14).
Diagnóstico diferencial (8
Defectos por transporte de nutrientes o electrolitos por diarreas congénitas cloradas, por sodio y malabsorción de glucosa-galactosa.
Defectos en el transporte epitelial y polaridad por enfermedad de inclusión de vellosidades y síndrome tricohepatoentérico.
A continuación, se hace referencia a otras enteropatías congénitas.
Diarrea congénita de sodio
En 1985, se describen trastornos diarreicos que se clasifican como diarrea sódica congénita (CSD, sigla en inglés) atribuída a defectos en la concentración de los iones de Na+ y H+. Posteriormente, Janecke y otros,34 identificaron mutaciones que dieron como resultado una reducción y/ o pérdida de la actividad de transporte basal de estos electrolitos.
Los pacientes afectados referían antecedentes de polihidramnios y diarrea posnatal asociada a deshidratación grave, pérdida de peso, hiponatremia y acidosis metabólica. Los desequilibrios electrolíticos en los casos más graves se asocian a un alto riesgo de mortalidad. Es una enfermedad rara con prevalencia menor a 1/ 1 000 000.34).
El sodio se cotransporta en el intestino, a través de la membrana del borde en cepillo mediante varias proteínas transportadoras específicas, como: 1) el cotransportador de sodio-glucosa, 2) los cotransportadores de aminoácido sódico, y 3) el intercambiador de sodio-hidrógeno. La forma clásica, del trastorno se debe a pérdida de la función del intercambiador intestinal de sodio/ hidrógeno 3 (NHE3).35,36).La función de este intercambiador intestinal es codificada por el gen SLC9A3 (también conocido como NHE-3), localizado en el brazo corto del cromosoma 5 (posición 15.33), lo que da como resultado una pérdida de la absorción de sodio y el bicarbonato, con secreción de líquidos y diarrea.
Se han identificado mutaciones causales tanto en SCL9A3 (herencia autosómica recesiva) como en el gen GUCY2C, localizado en el brazo corto del cromosoma 12 (12p12.3) (con herencia autosómica dominante), que codifican un receptor intestinal guanilatociclasa C (GC-C) y cuyas mutaciones activadoras inhiben NHE-3 (Cuadro 2). En 40 % de los pacientes aún no se ha identificado una mutación genética36,37).
El patrón de herencia es autosómico recesivo para las mutaciones SLC9A3 y autosómica dominante para las mutaciones GUCY2C (suelen tener un origen de novó). El riesgo de transmisión autosómico recesivo es de 25 %, cuando ambos padres son portadores no afectados. Hasta ahora, no hay información del mosaicismo de la línea germinal en los padres que puede resultar en riesgo de recurrencia muy bajo para los hermanos afectados. En varios individuos con mutaciones GUCY2C y SLC9A3 se ha detectado enfermedad inflamatoria intestinal.37,38).
El diagnóstico prenatal puede sospecharse ante polihidramnios en el tercer trimestre del embarazo, es posible realizar pruebas prenatales genéticas precoces y dirigidas después de identificar las variantes que causan la enfermedad en paciente índice.34,37,38
La diarrea de sodio congénita se caracteriza por deposiciones acuosas incesantes, de intensidad variable que se inicia desde el nacimiento independientemente de la alimentación. Se describe ausencia de expulsión de meconio e irritabilidad, en ocasiones apatía, deshidratación de moderada a severa asociada a distensión abdominal y oclusión intestinal secundaria a vólvulos, por la distensión de asas intestinales. Es una enfermedad rara y se han publicados menos de 50 casos en la literatura.39).
Las heces contienen cuantías altas de sodio (que puede ser normal cuando la depleción del sodio corporal ha progresado durante algún tiempo) y concentraciones bajas de sodio sérico, acidosis metabólica y pH fecal alcalino con excreción urinaria disminuida de sodio. Los hallazgos histológicos incluyen epitelio intestinal estructuralmente intacto y proporción normal criptas/ vellosidades. El diagnóstico debe confirmarse mediante pruebas genéticas.39).
El diagnóstico diferencial con la forma sindrómica de diarrea sódica congénita (debida a mutaciones en SPINT2), se distingue típicamente por atresia coanaluni o bilateral y características sindrómicas adicionales como dismorfismo facial, ano imperforado y manifestaciones oculares, como síndrome de ojo seco refractario a tratamiento, erosiones corneales y la presencia de coloboma ocular. Las mutaciones en el inhibidor de la serina proteasa, el gen Kunitz-Tipo 2 (SPINT2) también se ha asociado con diarrea congénita de sodio.36,40.41).Otros diagnósticos incluyen obstrucción intestinal anatómica, diarrea congénita por cloruro, malabsorción de glucosa-galactosa, enfermedad por inclusión de microvellosidades y enteropatía congénita por penacho.40,41).
El tratamiento es dependiente de NPT durante varios meses. Requieren suplementos de sodio y tratamiento de la deshidratación severa. Los suplementos de sodio y bicarbonato pueden permitir un crecimiento normal. Es una afección potencialmente mortal, en ciertos casos, los pacientes pueden dejar de recibir NPT, a largo plazo, mantienen una diarrea acuosa leve, retardo del crecimiento e hipoaldosteronismo.39).
Clorhidorrea congénita
La clorhidrorrea congénita se caracteriza por el antecedente de polihidramnios, es más frecuente en recién nacidos pretérminos, que, durante el primer año de vida, presentan trastornos nutricionales severos y deshidratación asociada a hipopotasemia, alcalosis metábolica, hipocloremia con pérdida excesiva de cloruros en las heces. En el reporte de caso realizado por Coronado42 y colaboradores se hace referencia a que está entidad fue descrita en 1945 por Gamble y Darrow que describen simultáneamente dos casos de niños con una forma inusual de diarrea severa asociada a una alcalosis metabólica en contraste con la acidosis habitual en estos cuadros. Más tarde, se establece el origen genético de la enfermedad42,43).
Es una enfermedad rara, que se debe a un defecto en el transporte activo Cl/ HCO3 mediado por SLC26A3 en el epitelio superficial del íleon y el colon, trasmitida de forma autosómica recesiva, por una mutación localizada en 7q3144 que da como resultado la pérdida de Cl en las heces y diarrea osmótica asociada a trastornos electrolíticos como la hipocloremia. Por otro lado, la acidez intestinal obstaculiza la absorción de sodio y agrava la hiponatremia. Se han publicado en la literatura alrededor de 250 pacientes y es más frecuente en países como Arabia Saudita, Kuwait, Finlandia y Polonia. En Cuba, se han informado en la literatura médica 2 niños con clorhidrorrea congénita. (43).
La alcalosis metabólica secundaria a inadecuada secreción de HCO3 en íleon y colon y excesiva pérdida de H+ a través de los riñones con hiperaldosteronismo secundario, es un mecanismo para conservar el sodio.42).
El cuadro clínico se caracteriza por antecedente de polihidramnios, parto pretérmino, comienzo inicial de diarrea osmótica, pérdida de cloro en las heces. Precozmente presenta distensión abdominal, asas delgadas con peristaltismo visible, diarreas acuosas y desnutrición asociado a retraso mental y sufre infecciones respiratorias. Se describe como complicaciones: vólvulos intestinales, infecciones urinarias a repetición y asociación a diversas alteraciones renales que incluyen insuficiencia renal crónica, nefrocalcinosis y síndrome nefrótico congénito. (42)
El diagnóstico se realiza por análisis electrolítico de heces con cloro aumentado, (alrededor de 150 meq/L), ausencia de cloro en orina, alcalosis hipoclorémica e hipopotasemia.40,41).En el periodo neonatal es sospechada por presencia de polihidramnios y dilatación significativa de asas intestinales. El estudio genético incluye la secuenciación del gen SLC26A3.44).
El tratamiento consiste en la sustitución de las pérdidas de agua y electrolitos para suplir las pérdidas de cloro y potasio. El total de la dosis de cloruro de sodio y cloruro de potasio se ajustará hasta la dosis mínima para mantener el pH sanguíneo y cubrir las pérdidas de cloro urinario, que permite un crecimiento y desarrollo normal del niño.42).
Malabsorción de glucosa galactosa
Es una enfermedad genética heredada de forma autosómica recesiva, causada por mutaciones gen SLC5A1, localizado en el cromosoma 22 (22q13.1), que regula el cotransportador de Na+/glucosa tipo 1, lo que trae como consecuencia que la glucosa y la galactosa no puedan ser absorbidas por el intestino. Se considera una enfermedad rara, se han comunicado mundialmente 300 casos y se conocen más de 40 mutaciones del gen45). (Cuadro 2).
La incidencia es variable según diferentes regiones geográficas y la presencia de consanguinidad es evidente en algunas áreas. (46,47.48)
La diarrea es acuosa severa, de inicio neonatal y lleva a la deshidratación, generalmente con hipernatremia. La secreción de agua y electrolitos en el yeyuno puede ser estimulada por la ingesta de sustancias osmóticas como la glucosa y la galactosa. Normalmente el agua pasa a través de la mucosa intestinal, con glucosa y galactosa, por la vía del tranportadorNa+/glucosa, pero en estos niños la glucosa y la galactosa no pueden ser absorbidas, por lo que grandes cantidades de agua, electrolitos y glucosa atraviesan el intestino delgado, exceden la capacidad de absorción del colon y se origina la diarrea acuosa, ácidas con alto contenido en glucosa, que conlleva a desnutrición, incluso a la muerte del paciente. La deshidratación frecuente puede determinar la aparición de daño renal como cálculos, poliuria, acidosis tubular renal y hematuria.46,47).
El diagnóstico puede confirmarse por cromatografía en heces. La realización de una prueba terapéutica con fórmula libre de carbohidratos resuelve la sintomatología rápidamente y mejora la evolución del paciente que normaliza los desequilibrios hidroelectrolíticos y aumenta progresivamente el peso. 46,47).
El tratamiento más efectivo es la eliminación de alimentos que contengan glucosa y galactosa de la dieta, incluso la leche materna, aunque a veces ha existido tolerancia. Este tratamiento produce resolución de los síntomas y desaparición de la desnutrición. Existen fórmulas alimentarias especiales libres de lactosa y ambos monosacáridos.46,47,48).
Enfermedad de inclusión de microvellosidades
Es una diarrea refractaria en el lactante, producida por alteración congénita del epitelio intestinal, que provoca diarrea acuosa abundante, asociada a malabsorción mantenida, que habitualmente determina dependencia de NPT durante toda la vida. Fue descrita en 1978 y su prevalencia se estima sea menor a 1:100 000 y se han descrito pocos casos en la literatura. Se hereda de forma autosómica recesiva, con leve predominio en el sexo femenino. Es frecuente el antecedente de consanguinidad y, generalmente no existen antecedentes prenatales, aunque se ha descrito oligohidramnios durante el embarazo.49).
La evolución crónica de la diarrea es causada por defectos estructurales de la mucosa, 50,51) por mutaciones en el gen MYO5B (18q) que da como resultado la alteración de la polaridad apical de las células epiteliales intestinales.52).La desorganización de las microvellosidades afecta la capacidad de absorción de los enterocitos. Una variante sin vesículas de inclusión microvellosa, es causada por mutaciones y pérdida de función en el gen de la sintaxina 3 (STX3) (11q12.1) y, cuando se asocia con linfohistiocitosis hemofagocítica, también puede deberse a mutación del gen STXBP2 (19p13.2). Algunos pacientes no presentan mutaciones en ningún gen identificado53) (Cuadro 2).
Las diarreas son acuosas severas, de tipo secretora con componente osmótico por el déficit de enzimas, en el borde en cepillo de las células epiteliales, con rápida deshidratación y desequilibrios hidroelectrolíticos. Existe una presentación neonatal precoz y una forma de aparición más tardía entre los 2 a 4 meses de vida.53,54). Hay retraso del desarrollo y se han notificado anomalías asociadas poco frecuentes (hernia inguinal, displasia renal). La NPT a largo plazo55 puede complicarse con colestasis hepática secundaria. También se han descrito formas atípicas, sin inclusiones microvellositarias detectables y de progresión menos grave. 53).
El diagnóstico es por microscopia óptica de mucosa del duodeno o yeyuno (tinción de hematoxilina y eosina) muestra atrofia hipoplásica variable de vellosidades y acumulación de gránulos secretores positivos a la tinción con ácido periódico de Schiff (PAS) en el citoplasma apical de los enterocitos, con ausencia de infiltrado inflamatorio. No hay alteraciones en las criptas. 46). La microscopia electrónica es fundamental para el diagnóstico, 8,15,56), muestra cambios patognomónicos, como vacuolas intracitoplasmáticas con microvellosidades incluidas y su ausencia o escasez en el borde luminal del epitelio intestinal, secundaria a la pérdida de la polarización de los enterocitos deficientes de MYO5.56). Las técnicas de inmunohistoquímica pueden mostrar una peptidasa neutra CD-10 dentro del citoplasma del enterocito y no en su superficie. 49). Las pruebas genéticas moleculares son esenciales para confirmar el diagnóstico. 53).
El tratamiento se basa en el trasplante de intestino, solo o en combinación con trasplante hepático. Habitualmente se logra sobrevida a base de NPT domiciliaria. En los pacientes trasplantados se ha descrito sobrevida de hasta en 70 % de los casos. Debido a la gravedad y la precocidad de los síntomas, así como a las complicaciones de la NPT a largo plazo, el pronóstico es generalmente desfavorable. Las causas de fallecimiento incluyen deshidratación grave y desequilibrio metabólico, desnutrición y sepsis.49,53).
Síndrome tricohepatoentérico
Es una afección rara y grave caracterizada por diarrea intratable con expresión sindrómica causada por mutaciones homocigóticas y heterocigóticas.57,58).Los pacientes con variantes patogénicas en TTC3759,) y SKIV2L60 presentan retraso del crecimiento intrauterino, anomalías del cabello “tricorexisnodosa” (espeso y lanoso, propenso a la rotura, en ocasiones escaso y fácil de arrancar); anomalías cutáneas (manchas color café con leche, xerosis y piel gomosa); dismorfia facial (frente y mejillas prominentes, raíz nasal amplia e hipertelorismo); afectación hepática (fibrosis extensa o cirrosis) e inmunodeficiencia, además puede presentarse con colitis.57,58),El mecanismo patogénico preciso no se conoce completamente, aunque se supone que es epitelial. La prevalencia es entre 1/300 000 a 1/400 000 nacidos vivos (57).
En general, el desarrollo científico-técnico en el ámbito de la salud y la enfermedad ha permitido desarrollar la comprensión de múltiples enfermedades, donde las enfermedades genéticas ocupan una posición destacada. Los registros nacionales creados de enfermedades raras y las ultrararas, estas últimas afectan< 5/100 000 habitantes, todas con una prevalencia diez veces menor en la población61) y, los grupos de trabajo dedicados a esta actividad creados en muchos países,62,63) organizados por especialidades médicas; resultan de gran interés para la investigación de las expresiones clínicas, imagenológicas, histológicas y, genéticas, en afecciones habitualmente desconocidas, con información documentada sobre 6172 enfermedades raras únicas, de las cuales 79,0 % son genéticas y el 69,9 % de inicio exclusivamente pediátricas con un predominio de prevalencia < 1/1 000 000.64).
En este contexto la DEI plantea la necesidad de NPT a largo plazo para mantener un estado nutricional adecuado ante aparición de fallo intestinal por complicaciones de insuficiencia continuada de la homeóstasis intestinal. Los casos más graves pueden requerir trasplante intestinal, como esperanza de vida y solución definitiva, aunque las limitaciones en la obtención del intestino a trasplantar y las posibles complicaciones por rechazo, representan riesgos severos de morbilidad y mortalidad. El desarrollo de esta técnica en centros altamente calificados ha aportado supervivencia y seguridad.
Conclusiones
Se actualizaron los criterios de la diarrea crónica intratable del lactante causada por enteropatías congénitas catalogadas como enfermedades raras, por defectos diferentes clasificados en cinco categorías fisiopatológicas. En este contexto, se describieron los aspectos patogénicos, clínicos, histológicos y genéticos vinculados a inducción de la dieta o transporte de electrolitos. Las denominadas enteropatías congénitas cursan con comienzo precoz después del nacimiento y causan malabsorción grave y mortalidad variable, son representativas de enfermedades de trascendencia en la infancia.
La displasia epitelial intestinal o enteropatía en penacho, es una de estas raras entidades, de herencia autosómica recesiva que incluye entre sus características atrofia severa de las vellosidades intestinales con hiperplasia de las criptas con las típicas vellosidades anormales por desorganización de los enterocitos superficiales en su porción apical, que adoptan una agrupación (apiñamiento) focal, que muestra forma de penacho. Se argumenta el diagnóstico clínico, genético e histológico, aunque en ocasiones puede mostrar modificaciones no concluyentes de la mucosa intestinal, al adoptar aspecto en parches. La gravedad de la sintomatología determina la necesidad de nutrición parenteral prolongada o permanente por la intolerancia a los alimentos, pérdida de peso, malabsorción y fallo de medro. El trasplante intestinal es una alternativa para el tratamiento del fallo intestinal y lograr esperanza de vida por recuperación de la homeostasis digestiva, aunque es una técnica desarrollada internacionalmente en limitados centros especializados.
Se describieron las diarreas congénitas por sodio y cloro, malabsorción de glucosa y galactosa, la enfermedad de inclusión de vellosidades y el síndrome tricohepatoentérico, raras enfermedades para el diagnóstico diferencial, incluido sus rasgos de diarreas intratables, mutaciones genéticas y el valor de la biopsia de duodeno para su identificación.

