










 Custom services
Custom services Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La displasia renal multiquística (DRMQ) o riñón multiquístico (RM) es una grave displasia descrita por Schwartz en 1936 en una pieza de nefrectomía de un niño de 7 meses,1 pero las características fundamentales de esta anomalía fueron detalladas por Spence en 1955, que destaca la importancia de separarla de las enfermedades poliquísticas y otras anomalías quísticas.2
Esta displasia que en el ultrasonido materno-fetal puede confundirse con una hidronefrosis y viceversa3,4,5 se caracteriza por la presencia de quistes no comunicantes, de tamaño variable, separados por parénquima displásico y la ausencia de un sistema calicial normal. Esta anomalía está asociada con atresia ureteral o ureteropélvica, el riñón afectado es afuncional o con muy escasa función (esencialmente afuncional), y la arteria renal ipsilateral está atrésica o hipoplásica.6,7,8 Es relativamente frecuente, y se estima que ocurre en 1 de 4000 a 1 en 4300 nacimientos.6,9
Para explicar su etiología o mecanismo de producción se han planteado dos hipótesis, pero las dos conducen a la atresia del uréter y cuando el riñón comienza a producir orina se producen dilataciones quísticas y el líquido que se acumula en el riñón, produce disrupción de la nefrogénesis y el parénquima renal no se desarrolla8,9. Esta atresia ureteral o ureteropélvica produce la DRMQ.
Se describen cuatro variedades de DRMQ. Cuando se produce atresia de uréter y pelvis se desarrolla la forma habitual, clásica o típica de DRMQ que produce la imagen en “racimo de uvas” en la ultrasonografía; cuando la atresia afecta el tercio superior del uréter se desarrolla una gran pelvis rodeada de pequeños quistes denominada forma hidronefrótica de DRMQ; cuando los quistes son muy pequeños -pueden ser incluso microscópicos, se desarrolla la variedad sólida de displasia y existe además una forma segmentaria de DRMQ. Esta última forma de displasia se produce cuando existe un doble sistema colector, y a su vez se describe como típica cuando la displasia afecta el segmento superior de un doble sistema; de otra forma se describe como atípica.10
Se debatió durante algún tiempo si la conducta debía ser quirúrgica (nefrectomía) o conservadora inicial. Antes del advenimiento del ultrasonido diagnóstico prenatal solo se diagnosticaban aquellas displasias que su tamaño hacía posible que la vista o la mano del médico pudieran detectarlas y la conducta era la extirpación de la displasia. El ultrasonido prenatal permite diagnosticar una DRMQ en el segundo o tercer trimestre del embarazo y esto ha hecho que la conducta quirúrgica haya cambiado. Hasta finales del pasado siglo se planteaba que debido a lo poco que se conocía sobre su desarrollo y complicaciones, el tratamiento de la DRMQ había sido controvertido y variaba desde la nefrectomía hasta la simple observación.11
Actualmente se sabe que la DRMQ unilateral por lo general tiene un buen pronóstico, que puede involucionar en breve tiempo, -incluso prenatalmente- y que las anomalías contralaterales son frecuentes, en primer lugar el reflujo vesicoureteral (RVU).12
La degeneración maligna es posible, pero se han descrito pocos casos.12. Los estudios y propuestas de Beckwith,13,14 han contribuido grandemente al criterio de tratamiento conservador. Nuestro objetivo es describir una paciente con tumor de Wilms asociado a una displasia renal multiquística.
Presentación del caso
Paciente de sexo femenino y 10 años de edad, que tres años antes presentó “dolorcito” en flanco izquierdo por lo que le realizaron ultrasonido abdominal y detectaron “quistes en el riñón izquierdo” (Fig. 1).
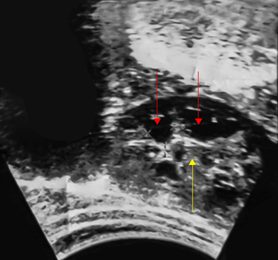
Fig. 1 Ultrasonido renal a los 7 años de edad donde se aprecia una displasia renal multiquística variedad sólida con quistes pequeños (flechas rojas) y predomino del estroma (flecha amarilla).
Por tal motivo, es remitida al servicio de urología donde es valorada y se cita dentro de 6 meses para su seguimiento, pero no regresa a consulta. Tres años después (agosto/2020) consulta por fiebre y síntomas respiratorios y se palpa tumoración abdominal por lo que es remitida. Se impone tratamiento para el proceso respiratorio y se comienza estudio de la tumoración abdominal. Al examen se palpa tumoración en flanco izquierdo de aproximadamente 4 cm de diámetro, superficie lisa, consistencia leñosa, no dolorosa y con contacto lumbar. Se hace ultrasonido abdominal donde se aprecia una masa sin aspecto reniforme con quistes de diferentes tamaños separados por la tumoración (Fig. 2); se discute colectivamente y se plantea realizar gammagrafía renal estática (DMSA) y predomina el criterio de realizar biopsia renal y nefrectomía izquierda. Se realiza biopsia renal percutánea y se informa nefroblastoma (tumor de Wilms). Se impone tratamiento citostático y se realiza nefrectomía.
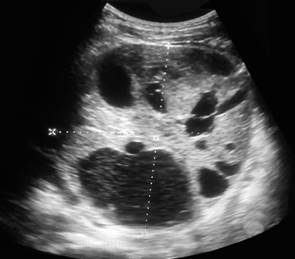
Fig. 2 Ultrasonido del riñón izquierdo a los 10 años de edad con quistes de diferentes tamaños dentro de una imagen tumoral.
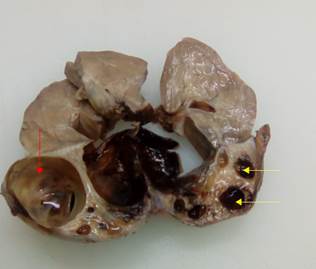
Por el resultado de la biopsia se impone tratamiento citostático con la finalidad de reducir la tumoración y se realiza nefrectomía. La pieza anatómica muestra una masa sin aspecto reniforme de 12,5 × 7,5 × 5,0 cm; se observa lesión predominantemente quística con el quiste mayor midiendo 5 cm de diámetro y otros quistes más pequeños. Se observa hemorragia escasa y necrosis de áreas sólidas, así como dilatación de cálices en polo opuesto al tumor y no se identifican los vasos del hilio renal (Fig. 3).
Discusión
La paciente que relatamos reúne tres elementos excepcionales: la posibilidad hereditaria por el hermano fallecido por DRMQ bilateral, un tumor maligno emergiendo de una DRMQ, y un tumor de Wilms a los 10 años de edad.
La DRMQ es una displasia no genética considerada como una anomalía del desarrollo, pero se han descrito casos familiares y se ha planteado una variedad con herencia autosómica dominante.15,16
Mutaciones del gen PAX2 -la causa del síndrome renal-coloboma- están asociadas con displasia renal. En tres generaciones de una familia se ha informado DRMQ que tenían mutación en este gen, sin alteraciones oculares, y estos hallazgos sugieren que todos los pacientes con malformaciones de riñón y tracto urinario pueden beneficiarse del estudio molecular de PAX2, señalan algunos autores.6,17 La paciente que presentamos nos permite pensar en esta posibilidad, sin poder afirmarla, por el hermano con DRMQ bilateral y la madre con RVU. La DRMQ bilateral se diagnostica en el 20 % de los casos detectados prenatalmente.7
Como se plantea en la introducción, la conducta médica para la atención de una displasia renal multiquística es muy discutida y en el momento actual existen criterios que recomiendan un tratamiento conservador pero con control clínico y ultrasonográfico períodico, que fue lo que no se cumplió en esta paciente que no regresó a consulta después de ser valorada clínica y ultrasonograficamente a los 7 años de edad.
Estors y otros,18 en 18 nefrectomías por riñones no funcionantes incluye 2 DRMQ por aumento de la masa renal y señalan que la DRMQ puede presentarse con masa renal de gran volumen con apariencia atípica o aumento de tamaño durante el seguimiento, en cuyo caso consideran la nefrectomía como el tratamiento de elección.
Entre las complicaciones que pueden presentarse en la DRMQ se citan el dolor en flanco, la hipertensión arterial, las infecciones urinarias y la degeneración maligna.6 En nuestra paciente el “dolorcito” en el flanco -un síntoma no frecuente en esta entidad- condujo al estudio ultrasonográfico que descubrió los quistes a los 7 años de edad. Multicystic Kidney Registry registró hipertensión arterial ligera en 4 (1,5 %) de 260 individuos con DRMQ que fueron observados al menos durante 5 años.19
Una revisión sistemática de 29 estudios con 1 115 niños con DRMQ y la probabilidad de desarrollo de hipertensión arterial en los niños con esta anomalía fue de 5,4 por 1 000.20
En otra revisión sistemática realizada en 2018, la incidencia de hipertensión arterial asociada a DRMQ fue 3,2 % (27/838),21 sin embargo, se ha planteado que la hipertensión arterial solo se presenta en esta anomalía si coexisten alteraciones del riñón contralateral.22
La mayoría de los sujetos con DRMQ no tienen flujo sanguíneo demostrable en el riñón displásico en los estudios radiológicos y un riñón exangüe afuncional es una causa improbable de hipertensión arterial; sin embargo, la presencia de anomalías congénitas contralaterales como la estenosis pieloureteral o la displasia renal contralateral, el desarrollo de cicatrices pospielonefríticas en el riñón con RVU o la hiperfiltración mantenida por el riñón funcionante son causas de hipertensión.6 Además se ha demostrado que en ocasiones la nefrectomía no resuelve la hipertensión.23
Otra complicación de la DRMQ es la infección del tracto urinario (ITU). Multicystic Kidney Registry informa ITU en 12 (5 %) en 260 niños con DRMQ.19 Pero se han registrado porcentajes mucho más altos (10/40).24
La pielonefritis, cuando se presenta, casi siempre es del riñón contralateral asociada a RVU y la infección del riñón multiquístico es rara porque la atresia ureteral, supuestamente, previene el ascenso del germen invasor,6 sin embargo, se ha comunicado la formación de absceso en el riñón displásico multiquístico25,26,27) y se ha descrito la ITU en el riñón con la displasia, presumiblemente, porque la bacteria infectante asciende traspasando la microluz del segmento atrésico del uréter.26
Otra complicación es la aparición de tumores malignos en el riñón con DRMQ, aunque se han publicado pocos casos.
Nunca se podrá decir con exactitud los casos que ocurren, algunos no se informan y otros pueden publicarse en idiomas que no leemos. Desde los estudios de Beckwith en la década de los años noventa del siglo xx,13,14 sabemos que esta complicación en el niño no es nada frecuente.
Wacksman y Sheldon en 1992,27 destacan que en los últimos 25 años solo se habían informado 6 casos de tumores malignos asociados a esta displasia, tres de ellos en niños.
Beckwith en 1992, mediante análisis matemático, no recomendó la nefrectomía por el cálculo de incidencia del tumor de Wilms,13 y cinco años después este mismo autor en el seguimiento de 7 500 casos de DRMQ estudiados durante más de 18 años donde solo 5 tenían esta asociación, señala que el tumor de Wilms es cuatro veces más frecuente en la DRMQ que en la población general.14
Beckwith apunta que el blastema nodular renal está presente en 1 % de la población general y que el tumor de Wilms se desarrolla en 1 de 8 000 niños. También señaló que aproximadamente 1 de 80 lactantes con blastema nodular renal desarrolla el tumor de Wilms.
La incidencia de blastema nodular renal es de aproximadamente 5 % en la DRMQ; por tanto, 20 riñones afectados con DRMQ sería necesario extirpar para quitar 1 con blastema nodular renal y 1 600 riñones con DRMQ tendríamos que remover para evitar 1 tumor de Wilms. Como la curación del tumor de Wilms es de aproximadamente 90 %, tendríamos que nefrectomizar 16 000 DRMQ para salvar una vida.13 Basado en estos datos Beckwith no recomienda la nefrectomía en la DRMQ.
Homsy y otros, en 199728 informan dos casos bien documentados de tumor de Wilms asociados a DRMQ en dos niñas de 5 y 3 meses, respectivamente. La de 5 meses fue seguida por diagnóstico prenatal de la DRMQ sin evidencia de tumor prenatal ni al nacer; la de 3 meses la llevan al especialista a consultarla por hipertensión arterial y defecto septal interventricular, el ultrasonido abdominal detectó el tumor.
Cui en 2010,29 comunica acerca de una niña de 5 años de edad con tumor rabdoide maligno asociado a DRMQ y señalan que es el primer caso de este tipo publicado en la literatura. Esa niña con diagnóstico prenatal de la DRMQ no tuvo un adecuado seguimiento posnatal y a los 5 años se detectó la tumoración al palparle el abdomen.
Rahman y otros, en 2000,23 por la descripción de 69 DRMQ unilaterales destacan que hasta 1997 se habían descrito 9 casos de tumores malignos con DRMQ (3 tumores de Wilms, 5 carcinomas y 1 mesotelioma), sin incluir las 2 pacientes con tumor de Wilms detectados por Homsy.28
Psooy en 2016, señala que entre 1983 y 1998 se describieron 5 casos de tumor de Wilms asociados a displasia renal multiquística en EE. UU., que representaron un riesgo de 0,03 a 0,1 % y que no se publicaron casos entre 1998 y 2007.30
Cambio y otros, revisaron artículos publicados en inglés entre 1965 y 2006 y destacan que el riesgo de tumor de Wilms es de 1 en 2 000 casos. También señalan que todos los casos se diagnostican antes de los 4 años, 70 % de ellos como una masa palpable y por último exponen que es práctica común la extirpación de la DRMQ palpable o que crece durante su evolución, pero que esto representa una fracción mínima.31
En una amplia revisión de DRMQ publicada en 2020,6 se señala que el nefroblastoma (tumor de Wilms) es la malignidad que con mayor frecuencia aparece en el niño con DRMQ, pero solo se han publicado 7 casos con edad promedio de 7 meses y refieren que se han informado 13 casos con carcinoma renal con edad promedio de 30 años y un tumor embrionario en un paciente de 68 años. No citan en esta revisión el tumor rabdoide maligno documentado por Cui y otros.29
Todos estos datos confirman que la paciente presentada es un caso raro, casi excepcional, que no hemos encontrado registrado en Cuba. Además confirman la necesidad de seguimiento prolongado en los pacientes con DRMQ unilateral, por lo que algunos plantean que la nefrectomía como conducta médica es controversial.32 Como las neoplasias son de origen estromal, no quísticas, aunque los quistes involucionen totalmente, la probabilidad del desarrollo de una neoplasia persiste.33
Concluimos que la paciente que se describe reúne tres características raras: un tumor maligno asociado a una displasia renal multiquística, la posibilidad hereditaria por el antecedente del hermano con la displasia bilateral y un tumor de Wilms a los 10 años de edad, lo que demuestra la importancia del seguimiento en estos pacientes.

