










 Custom services
Custom services Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La fibrosis quística (FQ) es un trastorno multisistémico, que afecta a niños y adultos, de evolución crónica, progresiva, y capaz de llevar a la muerte. Se trata de una enfermedad hereditaria autosómica recesiva, causada por mutaciones en ambas copias del gen de la proteína reguladora de la conducción transmembrana para la fibrosis quística, CFTR (por sus siglas en inglés: Cystic Fibrosis Transmembrane Conductance Regulator), la cual se sitúa en la superficie apical de las células epiteliales, y se comporta como una proteína transportadora multifuncional.1
Las mutaciones en CFTR provocan una falla en la conducción de agua, tanto en el interior como en el exterior de las células, lo que resulta en excedencia de secreción de moco anormalmente espeso y viscoso a nivel pulmonar, pancreático e intestinal, y que acarrea complicaciones serias e incluso la muerte.2
En el epitelio respiratorio, estas mutaciones determinan una alteración en las secreciones bronquiales, con aumento de su viscosidad y modificación de la depuración mucociliar. La infección endobronquial con microorganismos característicos, especialmente Pseudomona aeruginosa, induce un proceso inflamatorio persistente, no controlado, y se desencadena un círculo vicioso que conduce a la tríada característica de la enfermedad: obstrucción bronquial-inflamación-infección, que de seguir su evolución natural, conduce a daño pulmonar irreversible, con bronquiectasias, insuficiencia respiratoria y muerte.3,4,5
En la mayoría de los casos se encontró una o más expresiones clínicas de la enfermedad. Casi todos los pacientes exhibieron afección sinusopulmonar crónica, y entre el 85-90 % manifestó insuficiencia pancreática exocrina.6,7,8
En el diagnóstico de la enfermedad intervienen actualmente varias opciones: los diagnósticos prenatal y neonatal, el estudio genético, el test del sudor, y el estudio de la diferencia de potencial nasal.9,10,11,12
En Cuba, antes de implantarse el programa de pesquisa neonatal en todo el país, mediante la cuantificación de tripsina inmunorreactiva (TIR), en marzo de 2019, los diagnósticos se realizaban en casos con sospecha clínica, y se retardaba el establecimiento de un tratamiento precoz.
El pronóstico de la enfermedad evolucionó de forma favorable en los últimos años, de modo que la esperanza de vida de estos pacientes resultó algo superior a los 30 años.13,14,15 En ello influyeron múltiples factores, como son: el diagnóstico precoz, la aparición de nuevas terapéuticas para corregir la insuficiencia pancreática y enfrentar las infecciones respiratorias, el mantenimiento de una nutrición adecuada, y el tratamiento en centros especializados con un enfoque multidisciplinario.16,17,18,19
Para el abordaje de los múltiples aspectos de la enfermedad, el equipo de trabajo incluyó: neumólogo pediatra, gastroenterólogo, nutriólogo, genetista, fisiatra y psicólogo, además de otras especialidades que se consideraron necesarias, y la familia. El propósito fue tratar de forma beneficiosa a los enfermos conocidos, diagnosticar casos nuevos con el mayor rigor científico posible, identificar portadores y registrarlos, a fin de diagnosticar la enfermedad, y brindar asistencia psicosocial a pacientes y familiares.4,6,20
La provincia de Granma, brinda atención médica especializada hace casi 20 años a pacientes con FQ, en el Hospital Pediátrico Provincial Docente “Hermanos Cordové” de Manzanillo, Granma, pero son insuficientes los conocimientos sobre la morbilidad y mortalidad de estos pacientes.
El objetivo de este trabajo fue determinar la sobrevida de pacientes pediátricos con fibrosis quística hospitalizados en un centro especializado.
Métodos
Estudio observacional, de cohorte, retrospectivo, con todos los pacientes diagnosticados con FQ en el Hospital Pediátrico Provincial Docente “Hermanos Cordové” incluidos en el Registro Provincial de Fibrosis Quística, en el período comprendido entre abril de 2003 hasta abril de 2018. En este registro también se incluyeron, como parte del programa nacional de FQ, los pacientes que superaron la edad pediátrica.
El universo de estudio lo constituyeron 27 pacientes diagnosticados con FQ por sospecha clínica y confirmación mediante determinación de electrolitos en el sudor. A todo se les tomó muestra para estudio molecular.
Las variables estudiadas fueron: edad (en el momento del estudio), sexo, variantes genéticas de la enfermedad, colonización bacteriana y estado actual (vivo o fallecido). La supervivencia se consideró como los años transcurridos entre el diagnóstico e inicio del tratamiento y la edad actual o la edad al fallecimiento.
Las formas clínicas de la FQ se clasificaron como:21
FQ 1A: paciente con enfermedad pulmonar crónica de FQ y con IP (insuficiencia pancreática)
FQ 1B: paciente con enfermedad pulmonar de FQ sin IP.
FQ 2 (FQ atípica): paciente con cuadro respiratorio a repetición no típico (bronquitis crónica, sibilancia recurrente), pueden tener manifestaciones digestivas como IP, cuadros de deshidratación, entre otros.
FQ otras: pacientes prácticamente asintomáticos o con pocos síntomas, generalmente son hermanos de enfermos.
Se emplearon métodos empíricos y teóricos generales.
Se procedió a la revisión de la historia clínica de cada enfermo y se registraron los datos en una planilla de recolección de la información confeccionada para tal fin y que constituyó la fuente primaria del estudio.
Se empleó el paquete estadístico SPSS 25.0 (https://agetintopc.com/es/ibm-spss-statistics-25-free-download/) para el procesamiento de los datos a través de estadística descriptiva. Los resultados se expresaron en frecuencias absolutas y porcentajes.
Para definir los grupos de edades se utilizó la clasificación según el anuario estadístico de salud pública (lactante y preescolar, escolar, adolescencia temprana y pacientes con edad ≥ 15 años).3,12
La sobrevida se calculó desde la fecha de inicio del tratamiento hasta la muerte o última consulta. Las diferencias en sobrevida se determinaron mediante la prueba de log-Rank, y las curvas de sobrevida generadas, con el uso del método Kaplan-Meier (p< 0,05).
El estudio se aprobó por el Comité de Ëtica de la investigación y el Consejo Científico del Hospital Pediátrico Provincial Docente “Hermanos Cordové”. Se respetaron los principios de la bioética: justicia, beneficencia y no malevolencia. Se garantizó el anonimato y la confidencialidad de la información en todos los casos.
Resultados
En la muestra analizada predominó el sexo masculino con 17 (63 %) casos, y el grupo de edad ≥ 15 años, el de mayor representación. El paciente de mayor edad: 27 años. La razón de masculinidad (RM): de 1,8 por cada mujer (tabla 1).
Tabla 1 Características de los pacientes con fibrosis quística según edad y sexo
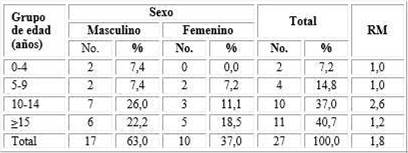
RM: razón de masculinidad
Predominó la variante F508del en 15 (55,5 %) pacientes, aunque de forma heterocigótica en 12 (44,4 %) de ellos En la tercera parte de los enfermos no se conoció la mutación genética debido a que no se estudiaban en Cuba (tabla 2).
Tabla 2 Variante genética en los pacientes diagnosticados con fibrosis quística
| Variantes genéticas | No. | % |
|---|---|---|
| F508del/ - | 12 | 44,4 |
| R 334 W/R 334 W | 2 | 7,4 |
| G5434/ - | 1 | 3,7 |
| F508del/F508del | 2 | 7,4 |
| F508del/G 5424 | 1 | 3,7 |
| No identificada | 9 | 33,3 |
| Total | 27 | 100,0 |
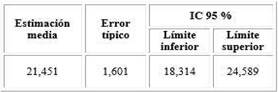
La supervivencia en 30 años del servicio resultó en 21,45 años (fig. 1 y tabla 3).
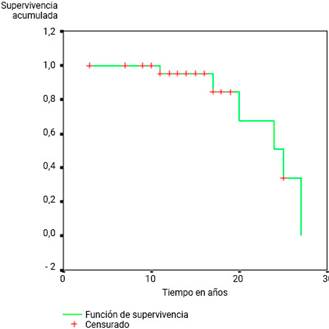
Fig. 1 Media de supervivencia de los pacientes con fibrosis quística desde el diagnóstico y comienzo del tratamiento de la enfermedad.
Se demostró una supervivencia mayor en el sexo femenino, pero sin significación estadística (p= 0,08) (fig. 2, tabla 4).
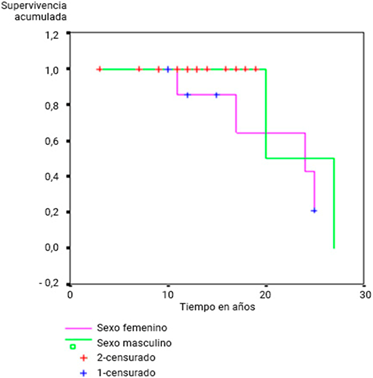
Tabla 4 Prueba de Mantel Cox. Comparación de las estimaciones de la función de riesgo.
| Gl | Significación | ||
|---|---|---|---|
| Log Rank (Mantel-Cox) | 2,240 | 1 | 0,134 |
Leyenda: GI = grado de libertad.
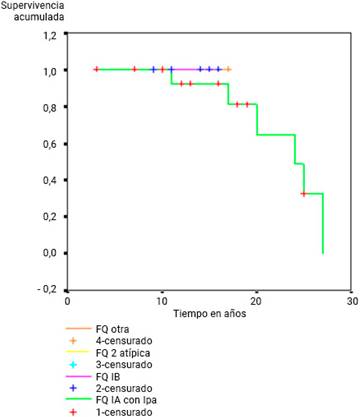
La FQ IA con IP, se manifestó como la forma clínica más severa. Se tomó como la de menor supervivencia, pero esta diferencia con el resto de las formas clínicas no resultó estadísticamente significativa (p= 0,07) (Fig. 3).
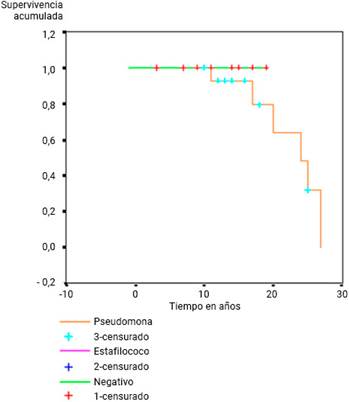
Los pacientes colonizados con Pseudomona aeruginosa mostraron una supervivencia menor, pero sin diferencias significativas entre las curvas de supervivencia (p= 0,08) (Fig. 4).
Discusión
La fibrosis quística se considera una enfermedad que afecta la calidad de vida y la supervivencia de los individuos. Esta última, en el momento actual, aumentó debido a la intervención precoz y agresiva ante las complicaciones, el tratamiento nutricional riguroso, y el desarrollo de centros con enfoque multidisciplinario, basados en algoritmos de tratamiento.21,22,23
En un estudio realizado en Chile se registró la mayor frecuencia de casos en el sexo masculino con 57,8 % en una serie de 48 niños evaluados en un período de 26 años;23 y en Cuba, en el Hospital Pediátrico Universitario "William Soler" de La Habana, en un espacio de 40 años, con 98 niños que representaron el 62,5 %. Este centro también informó mayor frecuencia de casos actuales en los niños de 15 o más años.12
En el Registro Nacional cubano, en el 2015, en general, predominó el sexo masculino en el 60 % de los casos,1) sin embargo, en Cienfuegos, en los 12 pacientes evaluados en consulta, en 2018, predominaron las niñas (66 %). Este centro también describió mayor número de enfermos con más de 18 años (33 %),3 cifra ligeramente inferior a la que se presenta en la presente investigación.3
Actualmente, se identificaron más de 1900 mutaciones diferentes en el gen CFTR, pero la deleción de la fenilalanina en la posición 508 (F508del) resultó la mutación más documentada,11 aunque su frecuencia varío de acuerdo con la población que se estudió: 48-50 % en España; 66 % en el Registro Nacional de Cuba; 47,8 % en México; 60,9 % en Argentina y 35,4 % en Brasil.1,4) Esta mutación, incluida en la clase II, provocó una alteración en el proceso postranscripcional de la proteína, con disminución del tráfico de la proteína madura desde el aparato de Golgi hacia la superficie celular apical.24,25
En el estudio realizado en Cienfuegos también predominó la mutación F508del, en 9 de los 12 casos, y mayor frecuencia de forma heterocigótica.3 En Cuba,11 las mutaciones G542X y R334W le siguieron en orden de frecuencia y se encontraron presentes en la serie de casos estudiada.
Aun cuando la tríada clínica clásica de la FQ comprendió la enfermedad pulmonar, de las glándulas sudoríparas y del páncreas, se consideró una afección multisistémica con efectos patológicos en una amplia gama de órganos que poseen tejido epitelial, e incluyó el tracto gastrointestinal.3,17,23
Se estimó que el 85 % de los pacientes con FQ presentaron insuficiencia pancreática, y se consideró que los factores genéticos influyeron en el grado de enfermedad pancreática y su ritmo de progreso. Se comprobó la existencia de una correlación grande con la mutación más común F508del, dado que 99 % de los casos homocigotos para dicha mutación presentaron insuficiencia pancreática.3,4,25 En la actual investigación, los resultados coincidieron con estos hallazgos.
En más de 90 % de los pacientes con FQ, el desenlace de la enfermedad obedeció a las complicaciones pulmonares, aunque pareció existir una asociación directa entre el grado de desnutrición y la gravedad de la enfermedad pulmonar; se reconoció que los pacientes con insuficiencia pancreática presentaron una menor supervivencia.5,21
A pesar de concebirse la FQ como enfermedad multisistémica, las manifestaciones respiratorias constituyeron la principal causa de morbilidad y mortalidad. Alrededor de la mitad de los niños con esta afección acudieron por primera vez a la consulta médica porque presentaron tos, respiración sibilante e infecciones del tracto respiratorio. Esta sintomatología resultó la más frecuente en los casos estudiados, con presencia de una enfermedad pulmonar típica, y coincidió con lo encontrado en otros estudios realizados en Cuba, en los que alcanzó hasta 40 % entre los enfermos.3,12,25
La infección respiratoria crónica persistente puede, en su inicio, resultar causada por Staphylococcus aureus, pero al final, colonizada por Pseudomona aeruginosa. En los casos estudiados el germen responsable de la mayor cantidad de infecciones respiratorias fue Pseudomona aeruginosa, al igual que lo encontrado en otros artículos publicados.12,23
La mortalidad por FQ resultó mayor entre las clases pobres, y entre los enfermos que se exponen al tabaquismo u otros polutantes. También aumentó cuando la infección por Pseudomona se cronificó debido a la no adherencia al tratamiento, o a la pobre ingestión de calorías que no cubrieron los requerimientos incrementados en estos pacientes.11,19
En un estudio realizado en Chile,26 período 1997-2003, 54,4 % de los pacientes fallecidos por FQ pertenecían al sexo femenino y el 66,0 % de la muestra estudiada falleció antes de los 15 años de edad. Este grupo de edad mostró el mayor número de defunciones en términos absolutos. Estos resultados correspondieron a un periodo anterior, en el que el arsenal terapéutico se encontraba menos desarrollado, lo que pudiera influir en la mortalidad. En la actualidad, el consenso chileno de 2020 registró una supervivencia promedio de 27 años,27) muy similar a la encontrada en pacientes granmenses.
Para Cuba, en el periodo 2001-2008, el promedio anual de fallecidos fue de 3,75; de ellos 33,3 % ocurrió en los menores de un año.28
La Sociedad Española de Neumología pediátrica describió una mediana de supervivencia de 41,6 años en 2015, en pacientes estadounidenses, según la Cystic Fibrosis Foundation en EE. UU.15
La Fundación Canadiense de Fibrosis Quística planteó en el 2016 una mediana de 51,8 años para sus enfermos.29 Esta mayor supervivencia se espera en países desarrollados, con todos los recursos disponibles y un nivel superior de vida.
En la investigación realizada en el Hospital “William Soler”, La Habana, Cuba,12 1977-2017, la mayoría de los fallecimientos ocurrieron antes del año 2000 con predominio de los menores de un año. En los últimos años del período analizado, 40,8 % de los asistentes a consulta resultaron adolescentes mayores de 15 años. Este resultado coincidió con lo encontrado en la presente investigación.
El estudio realizado en 4881 canadienses, 7329 franceses y 3896 personas australianas con FQ,30 arrojó, como resultados preliminares, que la mediana de edad general de supervivencia fue de 52,6 años para Canadá; 60,5 años para Francia y 53,3 años para Australia. Estos datos correspondieron a 2012 y 2016.
La mediana de edad de supervivencia en pacientes con FQ aumentó tanto en Canadá como en EE. UU. entre 1990 y 2013, sin embargo, en 1995 y 2005, la supervivencia en Canadá aumentó a un ritmo más rápido que en los Estados Unidos (p< 0,001). Sobre la base de datos contemporáneos, de 2009 a 2013, la mediana de edad de supervivencia en Canadá resultó 10 años mayor que en los EE. UU. (50,9 frente a 40,6 años, respectivamente).31
En un estudio realizado en Argentina32 entre el 2013 y el 2017, la mediana de edad de supervivencia fue de 50,5 años y la curva de supervivencia de las hembras resultó algo más baja que la de los varones. Aun Cuba queda muy por debajo de las estadísticas internacionales, como lo demostró el actual estudio en Granma, aunque existen regiones del país con resultados más satisfactorios como es La Habana.12
Cuba es un país de bajos recursos y bloqueado, pero presentó un trabajo sostenido en el seguimiento multidisciplinario de estos enfermos. El Estado cubano responde, de forma gratuita, con la obtención de los medicamentos fundamentales para el tratamiento de esta enfermedad, así como de un suplemento nutricional subsidiado, que garantiza el aporte nutricional necesario. Esto repercute en la supervivencia de estos pacientes.
Entre las limitaciones de este estudio se encuentran: no incluir algunos aspectos relacionados con la supervivencia como el estado nutricional, la edad al diagnóstico, el nivel de vida, la adherencia al tratamiento y asistencia regular a consulta, además de ser un estudio retrospectivo, pero sus resultados contribuirán a perfeccionar la atención a estos pacientes y resaltar la necesidad de un diagnóstico precoz y oportuno.
Se concluye que los pacientes con fibrosis quística en Granma, Cuba, mostraron un promedio de supervivencia de 21,4 años, inferior a lo documentado en países desarrollados. El Estado cubano garantiza recursos para la atención de estos pacientes, a pesar de las dificultades económicas del país.

