










 Custom services
Custom services Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Evans (SE) se define como la presencia de citopenias inmunes que afecta dos o más líneas celulares simultáneamente o secuencialmente. Generalmente se refiere a la combinación de anemia hemolítica autoinmune (AHAI) con trombocitopenia inmune primaria (TIP) pero puede incluir también neutropenia autoinmune (NAI). Su etiología se atribuye a la producción de autoanticuerpos (auto-Ac) patológicos contra las células sanguíneas pero su causa real se desconoce.
Se ha avanzado recientemente en el entendimiento de esta compleja entidad, revelando su frecuente asociación con alteraciones en la regulación inmune. No obstante, en la mayoría de los casos, el SE idiopático aún precede el diagnóstico de una enfermedad autoinmune o inmunodeficiencia.1
En este trabajo se explica la relación del SE con las alteraciones de la regulación del sistema inmune y las entidades asociadas más frecuentes.
Métodos
Se revisó la literatura especializada en los idiomas inglés y español a través del sitio web PubMed y el motor de búsqueda Google académico, de artículos publicados sobre SE y desregulación inmune. El 69,73 % correspondieron a los últimos 5 años. Se hizo un análisis y resumen de la bibliografía revisada.
Análisis y síntesis de la información
Incidencia
La verdadera incidencia del síndrome de Evans se desconoce dada la relativa rareza de esté, así como por las diferentes y versátiles definiciones a lo largo de los años. Las incidencias de TIP y AHAI individualmente se han establecido mejor y permite alguna estimación de la incidencia del SE.2,3
La trombocitopenia inmune primaria se diagnostica en 5-10 de cada 100.000 niños y 3,5 por 100.000 adultos por año,4,5 mientras que la AHAI se diagnostica en 1 a 3 de cada 100.000 niños y adultos por año.6 Los datos sitúan la aparición de SE entre los pacientes con TIP en el 1 %;7 aquellos con AHAI es más variable, entre 13-73 %.8 La incidencia de SE entre los pacientes con TIP crónica es más alta que entre el total de todos los pacientes con TIP, citada en 6,7 %.9 Los datos más recientes de incidencia SE, coinciden con los obtenidos entre los años 1960 a 1980, en que el diagnóstico de SE de todos los pacientes con TIP o AHAI como presentación inicial se halla entre 0,8 - 3,9 %.10
La edad promedio de presentación entre los niños es aproximadamente de 5 años.2 No existe predilección étnica o de género específica, aunque hay una tendencia al predominio masculino en pacientes jóvenes y al femenino en pacientes mayores.3
Hasta la fecha, la serie más grande ha demostrado que la AHAI y TIP ocurren simultáneamente durante la presentación inicial, en aproximadamente la mitad del tiempo (48 % en niños y 55 % en adultos).2,3 Muchos pacientes diagnosticados con SE presentarán inicialmente sólo una citopenia autoinmune: la TIP en 29 %, la AIHA en 24 % de los niños y 16 % de los adultos,11 y NAI en el 20 % de los niños2 y 15 % de los adultos.3
Los pacientes con síndrome de Evans presentarán síntomas relacionados con la/s citopenia/s autoinmunes de las cuales está compuesto su síndrome (anemia sintomática, ictericia, evidencia de hemólisis en AHAI, o petequias, hematomas y sangrado en la TIP). Sin embargo, a diferencia de las citopenias autoinmunes de linaje único de presentación aguda, el SE es a menudo una enfermedad crónica, recidivante y refractaria al tratamiento, secundaria a un trastorno subyacente de la regulación inmune. Por lo tanto, el diagnóstico de SE debe considerarse incluso al inicio de una citopenia autoinmune de linaje único, ya que las citopenias autoinmunes secuenciales ocurren con un promedio de 2,4 a 4,2 años después del diagnóstico de la citopenia autoinmune inicial, pero la presentación sintomática puede ser más grave.8,12
La mortalidad en el SE es entre 10 a 24 % en niños y adultos; por encima de la observada entre los pacientes con TIP, AHAI, o NAI solo y no está claramente relacionada con las complicaciones de la citopenia en todos los casos.2,3
Etiología
El síndrome de Evans se considera una condición "idiopática", sin embargo, los trastornos inmunitarios, ahora bien definidos, se identifican como la etiología de la enfermedad en aproximadamente el 50 % de los casos.
Las enfermedades más identificadas son las enfermedades autoinmunes sistémicas (EAS), con mayor frecuencia lupus eritematoso sistémico (LES), síndrome linfoproliferativo autoinmune (SLPA), y la inmunodeficiencia variable común (IDVC).1,13,14 Con el avance tecnológico y de las capacidades de diagnóstico, incluido un mayor acceso a los datos genómicos; las enfermedades inmunológicas hereditarias responsables de impulsar la autoinmunidad en el SE, se identificarán cada vez más.15
En la literatura se reportan casos con aberraciones inmunológicas, incluyendo linfadenopatía o hepatoesplenomegalia, hipogammaglobulinemia y citopenias autoinmunes, pero no cumplen los criterios para un diagnóstico de desregulación inmune.16 Estos casos no clasificados se comportan clínicamente de manera similar a los SE secundarios, que con mayor frecuencia requieren terapias de segunda línea (72 %) que los SE primarios (55 %).2 Esto resalta la importancia de la investigación de la inmunopatología subyacente no clasificada, ya que continuamente surgen diagnósticos novedosos y trastornos inmunes mejor definidos y se pueden descubrir mejores objetivos para terapias específicas.15
Enfermedades autoinmunes sistémicas
Las citopenias autoinmunes pueden ser una manifestación de presentación de EAS y se diagnostica aproximadamente en el 8 % de los pacientes pediátricos y 21 % de pacientes adultos con SE. Los pacientes que cumplen con los criterios de diagnóstico para una EAS, como el LES, ya no se clasifican con el diagnóstico de SE o SE secundario, ya que sus citopenias autoinmunes se consideran parte de su diagnóstico primario y deben tratarse en consecuencia.17
El lupus eritematoso sistémico es la enfermedad autoinmune más común diagnosticada en pacientes con SE, pero otras también pueden estar asociadas como: la tiroiditis de Hashimoto, la diabetes tipo 1, el síndrome de Ac antifosfolípidos y síndrome de Sjögren. Como mínimo, los pacientes con SE deben ser examinados para detectar LES.2,3,12,18)
Dadas las asociaciones anteriores, los niños y los adultos deben someterse a exámenes de detección de LES, SLPA e IDVC. En los adultos también se incluyen etiologías infecciosas como el virus de la inmunodeficiencia humana y el virus de la hepatitis C, además de tumores malignos, ya que se informan leucemias y linfomas en aproximadamente el 10 % de los adultos con SE (en comparación con 0 % en niños).3,12
Las investigaciones deben realizarse según esté clínicamente indicado, dada la amplia gama de entidades clínicas capaces de conducir al SE, incluidas infecciones, medicamentos, vacunas, inmunodeficiencias (inmunodeficiencias combinadas, síndrome de Di George, deficiencia selectiva de IgA), enfermedades autoinmunes, enfermedades linfoproliferativas, trastornos hemofagocíticos, neoplasias malignas y trastornos de la regulación inmune bien establecidos (inmunodesregulación poliendocrinopatía enteropatía ligada al X [síndrome IPEX]), como novedosos (deficiencia de CTLA-4 [del inglés cytotoxic T-lymphocyte antigen-4], deficiencia de LRBA (del inglés lipopolysaccharide-responsive beige-like anchor), síndrome PI3KD [del inglés activated phosphoinositide 3-kinase delta], el síndrome MonoMAC, entre otros).8)
Síndrome linfoproliferativo autoinmune
El síndrome linfoproliferativo autoinmune se define como linfoproliferación crónica no maligna, apoptosis linfocítica defectuosa mediada por Fas in vitro y elevado número de células T doble negativas TCRα/β+CD3+CD4−CD8− (DNT) en sangre periférica o tejido linfoide.18Teachey y otros, informaron por primera vez de la asociación de SLPA con SE idiopático, en el 50 % de los pacientes seguidos en su institución durante un período de 5 años.19 Posteriormente otro estudio confirmó los hallazgos previos en 47 % de pacientes pediátricos con SE que cumplían los criterios diagnósticos para SLPA.20
En el año 2009 se publicaron los criterios de diagnóstico revisados y la clasificación de SLPA con vistas a estandarizar el diagnóstico y modernizar la clasificación, y depender menos de técnicas obsoletas y engorrosas, y más fuertemente en las pruebas de células DNT altamente sensibles y específicas, y de técnicas específicas de mutación y variaciones de la enfermedad. Independientemente de estos matices, la fuerte asociación entre SLPA y SE es clara y probablemente se aclarará aún más en los próximos años.21,22
Inmunodeficiencia variable común
La prevalencia de citopenias autoinmunes en la IDVC es entre 10 - 20 % con incidencia de SE entre 3,4 - 3,8 %. Las citopenias autoinmunes parecen preceder al diagnóstico de IDVC en > 50 % de los casos y la incidencia en pacientes pediátricos con síndrome de Evans se informa en el 10 %. En adultos la incidencia de CVID es consistentemente más baja que la observada en pediatría, reportándose solo en el 6 %. Como regla general, SE está fuerte y frecuentemente asociado con IDVC, y todos los pacientes con SE deben ser evaluados para esta afección.23
Fisiopatología
Tradicionalmente, las citopenias autoinmunes en el síndrome de Evans, se han atribuido a la producción aberrante de auto-Ac y a la cascada de eventos posteriores que siguen, con una descripción más reciente de la actuación de la inmunidad celular en la patogénesis de la enfermedad. Sin embargo, en el 50 % de los casos se ha identificado únicamente una patología inmune subyacente, lo que permite una mejor comprensión de los verdaderos mecanismos de patogénesis en los casos de SE secundario.1
Enfermedades autoinmunes sistémicas
Las citopenias autoinmunes son una característica común de las EAS y, como tales, se incluyen en los criterios de clasificación y diagnóstico de estas enfermedades, en lugar de considerarse un fenómeno “secundario” de una entidad subyacente separada. Sin embargo, estas citopenias autoinmunes pueden ser una presentación inicial de la enfermedad y se consideran idiopáticas hasta que finalmente se diagnostica la enfermedad autoinmune primaria. Entre los pacientes con LES complicados por SE, el 50 % pueden ser diagnosticados con anomalías hematológicas (AHAI o TIP) meses o años antes de desarrollar manifestaciones clínicas de LES.24
La fisiopatología del síndrome de Evans en el espectro de estas enfermedades es diversa, pero específica de la enfermedad individual con la que está asociada. Una de las características más prominente es la incapacidad de inducir la tolerancia de los linfocitos y factores como la apoptosis defectuosa en las células B inmaduras, las señales de pro-supervivencia desequilibradas (las proporcionadas por BAFF [factor de activación de las células B]), la señalización alterada del receptor de las células B, que contribuyen al fracaso de la inducción de tolerancia central y periférica.23 La tolerancia defectuosa de las células T puede deberse a la incapacidad de generar células T reguladoras (Treg) o de anergizar las células T inmaduras que expresan receptores de células T autorreactivos.25
En el lupus eritematoso sistémico, la autoinmunidad humoral está implicada en la generación de citopenias autoinmunes con cambios inmunológicos que incluyen un aumento de precursores de células plasmáticas, expansión de células B CD19hiCD21lo/neg que parecen ser anérgicas pero expresan Ac autorreactivos de línea germinal y varios auto-Ac hematológicos específicos.26,27 Estos incluyen auto-Ac contra c-Mpl, o receptor de trombopoyetina,28,29 y Ac anti-GPIIb/IIIa en pacientes con TIP relacionada con LES, así como auto-Ac anti-CD40L, que se han identificado en pacientes con TIP y AHAI en el contexto del LES, pero no parecen expresarse en LES sin citopenias autoinmunes.30
El CD40L se expresa en la superficie de las células T CD4+ y plaquetas activadas, y se cree que estos auto-Ac desempeñan un papel en la desregulación de la tolerancia de los linfocitos y la patología de los complejos inmunes, así como en el aumento de la actividad antiplaquetaria.31 Además, la deficiencia del complemento que está bien descrita en el LES,15 que afecta más notablemente a C4, es más pronunciada en pacientes con TIP relacionada con LES y SE relacionada con LES en comparación con pacientes con TIP o SE o LES no complicados por citopenias autoinmunes.17,32
Síndrome linfoproliferativo autoinmune
El síndrome linfoproliferativo autoinmune es un síndrome en el cual la homeostasis de los linfocitos se interrumpe debido a mutaciones que afectan la vía apoptótica de Fas, lo que resulta en una supervivencia aberrante de linfocitos, linfoproliferación crónica, tolerancia inmune desregulada y autoinmunidad. El Fas expresado en la superficie de los linfocitos B y T activados y el ligando de Fas expresado en los linfocitos T activados interactúan para desencadenar la activación de la cascada de caspasas intracelulares que conduce a la apoptosis celular (Fig.).19,20,31
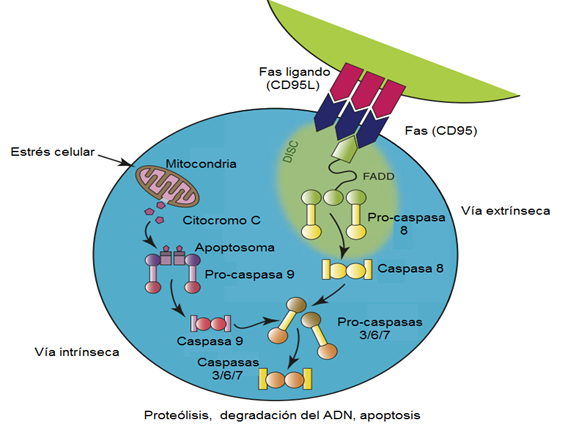 Fuente: Tomado y modificado de: Teachey DT, Lambert MP. Diagnosis and management of autoimmune cytopenias in childhood. Pediatr Clin North Am. 2013;60(6):1489-511).31
Fuente: Tomado y modificado de: Teachey DT, Lambert MP. Diagnosis and management of autoimmune cytopenias in childhood. Pediatr Clin North Am. 2013;60(6):1489-511).31
Fig. 1 Apoptosis celular.
Esta vía apoptótica funciona para regular negativamente la respuesta inmune al eliminar el exceso de linfocitos activados y autorreactivos, pero se vuelve disfuncional por mutaciones en los genes FAS (60-70%), FASL (<1%) y CASP10 (2-3%). Esto da como resultado la persistencia de células DNT características en esta afección, así como el fenotipo clínico característico de la linfoproliferación crónica y la autoinmunidad.33
La apoptosis defectuosa mediada por Fas se describe como una interrupción en la homeostasis de los linfocitos y otras interrupciones posteriores del equilibrio inmunológico que explican tanto la autoinmunidad mediada por células como la humoral observada en SLPA. Hay una linfocitosis de células B y T anormalmente persistentes y activados, tanto en sangre periférica como en los tejidos. Estos promueven un medio inmunitario que regula negativamente las respuestas de T-helper 1 (Th1) y aumenta las respuestas de T-helper 2 (Th2), con niveles altos de interleucina-10 (IL-10) y la disminución de los niveles de citocinas Th1.34
La fuente de producción de IL-10 no está del todo clara, aunque se ha demostrado que los monocitos y macrófagos de pacientes con SLPA producen 5 veces más IL-10 que los controles y existe la evidencia de la expresión potencialmente constitutiva de IL-10 por la población de células DNT en pacientes con SLPA. La IL-10 impulsa la diferenciación de las células T hacia un fenotipo Th2, e induce la proteína antiapoptótica Bcl-2 en células B y T, provocando una mayor persistencia patológica y expansión de los linfocitos autorreactivos. Las citocinas Th2 estimulan las interacciones de las células T y B necesarias para la producción de Ac, lo que explica la producción excesiva de auto-Ac.34,35
Los fenotipos clínicos e inmunobiológicos varían dentro del SLPA dada la variabilidad genética de la enfermedad, para lo cual se instituyó un sistema de clasificación basado en los genes que son afectados por mutaciones. La categoría más grande, "ALPS-FAS", abarca la enfermedad causada por mutaciones de línea germinal en FAS, que generalmente se heredan de forma autosómica dominante y se presentan en el 60 - 70 % de los pacientes afectados. En general, se trata de mutaciones heterocigóticas dominantes, que afectan el dominio intracelular del dominio de muerte activada por Fas. Sin embargo, el 30 % de las mutaciones de FAS afectan el dominio extracelular del dominio de muerte y, en su lugar, producen haploinsuficiencia. A diferencia de las mutaciones intracelulares dominantes, que generalmente resultan en ausencia de actividad Fas y mayor penetrancia de la enfermedad, estas mutaciones extracelulares haploinsuficientes generalmente conducen solo a una disminución de la actividad Fas, lo que resulta en una menor penetrancia de la enfermedad.35,36
Las mutaciones somáticas de Fas (ALPS-sFAS) ocurren en el 10 % de los pacientes y se limitan al compartimento de las células DNT, pero producen un fenotipo clínico similar al de los pacientes con mutación de línea germinal, excepto que los pacientes con mutaciones de línea germinal generalmente presentan SLPA a una edad más temprana. Todavía puede ocurrir algún cruce entre las dos categorías de mutaciones de FAS (ALPS-FAS y ALPS-sFAS), ya que las mutaciones de FAS tienen penetrancia variable, y algunas mutaciones requieren un segundo "golpe" para provocar una enfermedad clínica evidente. Como ejemplo, se ha identificado un pequeño subconjunto de pacientes con ALPS con múltiples mutaciones FAS, en quienes una mutación heterocigótica primaria de línea germinal no produce evidencia clínica de enfermedad, pero una mutación FAS somática que afecta el segundo alelo produce enfermedad clínica.33,37
Se han descrito otros fenotipos SLPA, en los que una mutación del gen FAS y una segunda mutación en el gen PRF1 (perforina) o el gen CASP10; y los miembros de la familia que portaban solo una de las mutaciones no tenía características clínicas de la enfermedad. Las categorías restantes utilizadas para clasificar ALPS son "ALPS-FASL", que abarca < 1 % de los pacientes con mutaciones FASL de línea germinal; "ALPS-CASP10", que abarca del 2 - 3 % de los pacientes con mutaciones en línea germinal de CASP10; y "ALPS-U", que incluye el 20 - 30 % restante de pacientes, en los que aún no se ha identificado ninguna mutación.33,38
Inmunodeficiencia variable común
La inmunodeficiencia variable común es el resultado de múltiples factores genéticos, ambientales e inmunológicos, ya que no se ha identificado una única mutación causante o vía de mutaciones.39 Las características de la enfermedad (hipogammaglobulinemia y respuestas de Ac deterioradas) proporcionan una justificación clara de las complicaciones infecciosas de esta afección. Sin embargo, como la terapia de reemplazo de inmunoglobulina se ha convertido en una práctica común, las complicaciones infecciosas han sido superadas por las complicaciones autoinmunes y linfoproliferativas como linfoproliferación, enfermedad granulomatosa, linfoma, enfermedad hepática, enteropatía y enfermedad pulmonar intersticial, granular o nodular. Este fenotipo compartido de autoinmunidad y complicaciones linfoproliferativas sugiere una inmunopatología subyacente común.40,41
La patología de la IDVC es diversa y multifacética, la cual es el resultado de la maduración deteriorada de células B y otros mecanismos inmunes complejos. Por ejemplo, se han observado niveles reducidos de células Treg en la IDVC complicada por autoinmunidad. Sin embargo, el desarrollo de linfocitos B sigue siendo clave para la patología de la enfermedad, ya que la maduración alterada de células B puede dar como resultado vías alteradas de señalización del receptor de células B, y finalmente, falla de los puntos de control de tolerancia central.42) Las células B autorreactivas normalmente seleccionadas para la edición del receptor o la anergia clonal evadirán la detección y escaparán a la periferia sin control.43)
Por otra parte, la disminución de los niveles de células B de memoria con cambio de isotipo, sugiere un desarrollo defectuoso del centro germinal, y una mayor preservación de la producción de IgM, lo que sugiere defectos potenciales en la participación de células T en el cambio de inmunoglobulina.41 Estas asociaciones se han fortalecido aún más por el descubrimiento de mutaciones en el gen TNFRSF13B que codifica el miembro de la familia del receptor del factor de necrosis tumoral TACI (activador transmembrana y modulador de calcio y receptor del ligando de ciclofilina), que media el cambio de isotipo en las células B.44 Estas mutaciones están relacionadas con el desarrollo de IDVC, con una alta asociación de complicaciones autoinmunes y linfoproliferativas. Además, se ha identificado la expansión de ciertas poblaciones de células B como las células CD19hiCD21lo/neg, que parecen expresar Ac autorreactivos.27)
Nuevos trastornos de la desregulación inmune y su relación con el síndrome de Evans
Actualmente, se han descrito varios síndromes similares a SLPA y síndromes de superposición SLPA-IDVC, en los que fenotipos clínicos que incluyen citopenias autoinmunes multilinaje que pueden identificarse inicialmente como SE indican una desregulación inmunitaria subyacente similar. Estos síndromes ahora se definen mejor y se reconocen cada vez más entre esta población de pacientes.45
El sistema de clasificación para SLPA no solo proporciona subclasificaciones para SLPA, sino que también proporciona un sistema de clasificación para los trastornos relacionados. Esto incluye la enfermedad leucoproliferativa autoinmune asociada a RAS (ELAR), asociada con mutaciones somáticas en los genes NRAS o KRAS;46,47 enfermedad linfoproliferativa autoinmune de Dianzani (ELAD), en la que aún no se ha identificado una mutación genética asociada; y otras clases en las que la linfoproliferación y la inmunodeficiencia, más que la autoinmunidad, son características más prominentes, incluido el estado de deficiencia de caspasa 8 (CEDS) y el síndrome linfoproliferativo ligado al cromosoma X (SLPX1).22,48
La ELAR se define clínicamente por citopenias autoinmunes, monocitosis y linfoproliferación, pero puede o no cumplir con todos los criterios de diagnóstico para SLPA o exhibir una elevación típica de IL-10, FASL soluble o altos niveles de vitamina B12, aunque la característica típica del SLPA como elevado número de células DNT parece estar presente con más frecuencia en la variante mutada de NRAS que en la variante mutada en KRAS; y la apoptosis defectuosa que se observa en ELAR no está mediada por Fas.49,50
Los genes RAS (incluidos NRAS y KRAS) codifican pequeñas proteínas de señalización intracelular de unión a GTP que funcionan en variedad de funciones, incluida la proliferación celular, el crecimiento y la apoptosis.49 Las mutaciones que activan la línea germinal en NRAS aumentan la señalización de RAF/MEK/ERK, que disminuye el mediador de la muerte celular (BIM) que interactúa con la proteína proapoptótica Bcl-2 y mitiga la apoptosis intrínseca inducida por la abstinencia de citocinas. De manera similar, la activación de mutaciones de KRAS da como resultado una apoptosis intrínseca alterada de células T a través de la supresión de BIM y pueden acelerar la proliferación celular a través de la represión de la proteína inhibidora del ciclo celular p27kip1.48
La ELAD se define clínicamente por la autoinmunidad e inmunodeficiencia típicas de SLPA, pero se distingue porque las poblaciones de células DNT a menudo no están elevadas y, a pesar de la evidencia de apoptosis defectuosa mediada por Fas, no hay mutaciones del gen FAS. La apoptosis defectuosa aparece, al menos en parte, debido a la alteración de la muerte celular inducida por ceramida, lo que indica que la ELAD puede ser causada por alteraciones posteriores en la vía de señalización de Fas.50
Algunos de los síndromes similares a SLPA descritos recientemente con características autoinmunes destacadas son la deficiencia CTLA-4,51 deficiencia de proteína de anclaje tipo beige que responde a lipopolisacáridos (LRBA),52,53 síndrome de fosfoinositido 3-quinasa δ activado (PI3KD),54 y mutaciones en la función del transductor de señal y activador de la transcripción (STAT).55
El CTLA-4 es un importante regulador negativo de las respuestas inmunitarias, y las mutaciones de CTLA-4 dan como resultado una unión alterada del ligando o una haploinsuficiencia de CTLA-4 y como resultado células TregFoxP3+ desreguladas, hiperactivación de las células T efectoras, pérdida progresiva de células B circulantes (con un aumento de células CD21lo/neg predominantemente autorreactivas), infiltraciones de órganos tanto linfocitos T como B, y alteración generalizada de la homeostasis de los linfocitos.56,57 Clínicamente, la haploinsuficiencia de CTLA-4 se caracteriza por linfoproliferación, infiltración linfocítica de órganos no linfoides, citopenias autoinmunes, hipogammaglobulinemia e infecciones recurrentes.51
La deficiencia de LRBA se caracteriza por un fenotipo clínico grave, con hipogammaglobulinemia y autoinmunidad de inicio temprano, con AHAI e TIP en > 50 % de los casos y también una fuerte asociación con la enfermedad inflamatoria intestinal. Las mutaciones homocigotas y heterocigotas compuestas de LRBA dan como resultado una enfermedad clínicamente relevante, mientras que los portadores de mutaciones heterocigotas parecen no verse clínicamente afectados.58,59
Aunque la función de LRBA en la respuesta inmune no está del todo clara, en su deficiencia existe interrupción en el desarrollo de células B, con defectos en la diferenciación, autofagia defectuosa, disminución de plasmablastos y células B de memoria, respuestas de Ac deterioradas e hipogammaglobulinemia; con alteración de la homeostasis de las células T dada la disminución de las células Treg observada en estos pacientes.52)
Los investigadores plantean que LRBA puede regular el inhibidor de CTLA-4, lo que explica el fenotipo inmunitario similar de la enfermedad que se observa en la haploinsuficiencia de CTLA-4 y la deficiencia de LRBA. Se están realizando más investigaciones sobre la verdadera patogenia subyacente a la deficiencia de LRBA, pero la alteración significativa en la homeostasis de los linfocitos y el desarrollo de las células B claramente impulsa una enfermedad clínicamente grave, que debe sospecharse y diagnosticarse al principio de su curso.60
El síndrome de PI3KD es el resultado de mutaciones en el gen que codifica la subunidad catalítica de PI(3)K p110δ. Esta subunidad catalítica se expresa selectivamente en leucocitos; y aunque la comprensión de su inmunobiología no está del todo clara, se sabe que tiene una función importante en la inmunidad adaptativa, al menos en parte a través de su papel en la activación de la quinasa diana de rapamicina (mTOR) en mamíferos.61,62 Además de activar mTOR, PI3K activa Akt (proteína quinasa B), que promueve la traducción de proteínas, incluida la proteína ribosómica 6s, que se combinan para promover la diferenciación de células T vírgenes en células T efectoras.62,63,64,65
La vía PI3K-Akt-mTOR activada constitutivamente en el síndrome PI3KD parece resultar en una deficiencia de células T vírgenes y una diferenciación sesgada de células T CD8+ a células T efectoras de vida corta y luego senescentes, con un desarrollo de células T y B de memoria gravemente deteriorado. La desregulación inmunitaria resultante se caracteriza clínicamente por complicaciones linfoproliferativas e infecciosas (incluidas infecciones sinopulmonares recurrentes y viremia por CMV y EBV), pero > 1/3 de los pacientes presentan autoinmunidad, incluidas citopenias autoinmunes multilinaje.66
Las mutaciones en los genes STAT, tanto en STAT1 como en STAT3, dan como resultado fenotipos clínicos caracterizados por inmunodeficiencia, susceptibilidad infecciosa y autoinmunidad.67 La fosforilación sostenida de STAT1 en mutaciones de ganancia de función de STAT1 da como resultado una diferenciación celular TH17 alterada y respuestas exageradas a la estimulación con interferón. Estas respuestas de células T auxiliares desreguladas generalmente dan como resultado un fenotipo diverso de inmunodeficiencia (incluida la candidiasis mucocutánea crónica) y autoinmunidad.68 De manera similar, las mutaciones de ganancia de función de STAT3 (incluidas las mutaciones somáticas y de línea germinal) producen diversos fenotipos autoinmunitarios y linfoproliferativos, pero las mutaciones de línea germinal dan como resultado específicamente un síndrome similar a ALPS con linfoproliferación, inmunodeficiencia y autoinmunidad, con autoinmunidad hematológica como principal.69 En particular, en estos trastornos se han identificado características similares a ALPS que incluyen poblaciones elevadas de células DNT y apoptosis defectuosa mediada por Fas, y se ha observado un deterioro de la apoptosis intrínseca, a través del aumento de proteínas antiapoptóticas Bcl-2. El aumento de la actividad de STAT3 produce otros efectos de señalización descendente con una disminución de la fosforilación de STAT1 y STAT5 en respuesta a IFN-γ e IL-2 y una disminución de las poblaciones de células Treg.70,71
Los síndromes en los que las características de SLPA y IDVC se superponen se identifican cada vez más. Si bien la hipergammaglobulinemia se informa en la mayoría de los pacientes con SLPA, a diferencia de la hipogammaglobulinemia definitiva de la IDVC, existen muchas características inmunopatológicas superpuestas que ocurren en estos dos síndromes.72)
Alteraciones similares de la diferenciación de células B, sobre todo niveles reducidos de CD27+, elevada población de células DNT y la apoptosis defectuosa mediada por Fas que es típica de SLPA se han identificado en varios casos de IDVC; y existe una clara evidencia de que Fas juega un papel importante en la maduración adecuada de las células B. Al mismo tiempo, se ha identificado hipogammaglobulinemia mucho más típica de IDVC en un subconjunto de pacientes con SLPA.73,74 Estos casos de “superposición” se complican por citopenias autoinmunes, a menudo multilinaje, en > 85 % de los casos. Aunque se han identificado pocas mutaciones en el gen FAS entre estos casos, la mayoría permanece sin clasificar genéticamente, pero es clínica e inmunofenotípicamente distinta, lo que genera la hipótesis de que estos síndromes de superposición SLPA-IDVC probablemente sean el resultado de combinaciones variables de mutaciones que involucran dos o más genes, aunque de alguna manera esté potencialmente mediado por Fas.75
Consideraciones finales
El síndrome de Evans es una condición en la que ocurren dos o más citopenias autoinmunes en un paciente, ya sea de forma simultánea o secuencial. El curso de la enfermedad es generalmente más crónico, grave y refractario al tratamiento que en las citopenias autoinmunes aisladas debido a la compleja desregulación inmunitaria subyacente. La inmunopatología, generalmente, se puede atribuir a una alteración en el desarrollo o la función de los linfocitos, de manera que el equilibrio inmunológico se inclina hacia la autorreactividad. En este momento sigue siendo un diagnóstico de exclusión, y se debe completar una investigación exhaustiva de las etiologías subyacentes antes de hacer este diagnóstico.

