










 Custom services
Custom services Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
Los Hidrocarburos Aromáticos Policíclicos (HAPs) son moléculas de dos o más anillos aromáticos fusionados de reconocida capacidad carcinogénica. Su presencia en la atmósfera, suelo, sedimentos, plantas y animales marinos hace que la exposición del hombre a los mismos sea inevitable.1 La contaminación por HAPs en su mayoría, tiene un origen antropogénico, estos se originan de la combustión incompleta o pirólisis de la materia orgánica. Una vez liberados en la zona de combustión en forma de vapores, debido a sus bajas presiones de vapor, se condensan sobre partículas de hollín.2
Pueden ingresar al ecosistema marino a través de la atmósfera, descargas o vertidos directos, mediante lixiviado de suelos circundantes, deposición atmosférica y por biosíntesis.3 Dentro de los ambientes acuáticos, los HAPs sufren una serie de procesos de intemperización (físicos, químicos y biológicos), mediante los cuales se asocian fácilmente con la materia particulada y finalmente se depositan en el sedimento.4 Como consecuencia, los sedimentos actúan como reservorios o registros de estos contaminantes. Además, por su persistencia natural a la degradación, los HAPs permanecen invariables como testigos de los procesos que le dieron origen.5,6
Las ventajas de analizar los sedimentos como matriz ambiental, en lugar de la biota o el agua, para explicar el comportamiento de estos contaminantes es que los concentran por períodos más largos. Además, los sedimentos son estáticos, su análisis es más simple, son relativamente fáciles de colectar y de extraer los analitos de interés, así como para repetir los análisis en los mismos sitios de interés, lo que permite estudiar las tendencias de la contaminación por períodos de tiempo específicos. El estudio de los sedimentos también puede aportar información sobre áreas con tendencia a la acumulación de contaminantes y brindar datos del estado previo a un evento antropogénico, es decir, contienen el registro del nivel base de compuestos de interés, su variabilidad y la influencia que tienen en un ecosistema.7,8
Existen varias técnicas para extraer HAPs desde sedimentos.1,9-10 Una de ellas implica someter la muestra a un tratamiento con ultrasonidos, mediante inmersión en un baño que produce una cavitación del disolvente alrededor de las partículas de la matriz, aumentando el contacto con el disolvente y mezclándolo con la muestra. Normalmente, para muestras granulares como suelos y sedimentos 11 se utilizan como disolventes el cloruro de metileno, n-hexano y acetona. Esta técnica permite la extracción de un número considerable de muestras con un costo relativamente bajo.12 Con este método de extracción se obtienen recuperaciones comparables a otros procedimientos convencionales, para determinados analitos y a partir de diversas matrices 9, aunque normalmente no son superiores a las obtenidas utilizando el método Soxhlet.13
La mayor parte de los HAPs pueden ser cuantificados por cromatografía de gases (GC), incluidos aquellos que han sido clasificados por la Agencia de Protección Ambiental de los Estados Unidos (US EPA) como contaminantes prioritarios.14 Los detectores más empleados para el análisis de HAPs por GC son: ionización por llama (Flame Ionization Detector, FID) 15 y principalmente el de espectrometría de masas (Mass Spectrometry, MS).16-17
El presente trabajo tiene como propósito implementar un método analítico que incluya metodologías de separación y concentración para identificar y cuantificar los 15 (HAPs) reconocidos como contaminantes prioritarios en sedimentos. Para ello es necesario asimilar el método estandarizado US EPA 8100 para la determinación de (HAPs), que utiliza la extracción asistida con ultrasonidos y la determinación por Cromatografía de Gases. La asimilación de dicho método requirió someterlo a un proceso de verificación interna, de acuerdo con la normativa cubana vigente, para lo cual se empleó un sedimento de referencia certificado. Finalmente se estudió la contaminación por HAPs en muestras de sedimentos del embalse Hanabanilla y la bahía de Cienfuegos.
Materiales y métodos
Equipos y materiales
Las determinaciones se realizaron en un cromatógrafo de gases marca SHIMADZU GC-2007, con autoinyector SHIMADZU AOC-20i y detector de ionización por llama. Para las extracciones se utilizó un baño ultrasónico SONOREX RK 52 (Berlín, Germany). Toda la cristalería y el equipamiento utilizado en el análisis fueron verificados y calibrados por la Oficina Territorial de Normalización de Villa Clara (OTN). Los reactivos empleados fueron de calidad cromatográfica, así como los disolventes empleados de calidad HPLC. Las disoluciones de referencia se prepararon a partir de un ámpula certificada (SUPELCO, U-97940. Supelco Analytical) de concentración 10µg/mL de cada uno de los HAP en estudio. También se dispuso de un sedimento certificado (IAEA - 417) 18 proporcionado por la Agencia Internacional de Energía Atómica.
Extracción de las muestras de sedimentos
El método estandarizado US EPA 8100 consta de cinco etapas.15 La primera es el pretratamiento de la muestra y consta de los procesos de secado y tamizado, lo cual se realizó siguiendo los estándares del método US EPA 610.19 La etapa de extracción asistida con ultrasonido se llevó a cabo teniendo en cuenta el procedimiento US EPA 3550C.20 La purificación del extracto se realizó de acuerdo con la metodología descrita en el método US EPA 3630ª.21
Análisis de los extractos por Cromatografía de Gases
Para la determinación de los HAPs por GC se empleó un cromatógrafo (SHIMADZU 2010) con detector de ionización por llama (FID), columna capilar SH-RtxTM-5 de 30 m x 0,25 mm ID. x 0,25 μm, nitrógeno como gas portador, a un flujo de 50 mL/min y una velocidad de flujo de 25,6 cm/segundo. Se utiliza un autoinyector automatizado, con inyección en modo splittless o sin división de flujo. Para la separación cromatográfica se estableció el programa de temperatura siguiente: 80 ºC por 5 min, incremento a razón de 8,4 ºC/min hasta 300 ºC, mantener a 300 ºC por 20 min.
Cálculo y expresión de los resultados
El contenido de cada HAPs en µg/g, se determina de la siguiente manera:
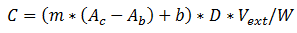 (1)
(1)donde C es la concentración de analito (en ng/g) m es la pendiente de la curva de calibración correspondiente b es el intercepto de la curva de calibración Ac: Área de la señal del analito en el cromatograma, en µV, Ab es el área de la señal del blanco, en µV Vext: Volumen final del extracto, en mL D es el factor de dilución W es el peso de la muestra inicial, en g
Verificación del método
Para seleccionar los parámetros de calidad del método analítico que deben ser evaluados, se procedió según la Norma Cubana Especificación Técnica TS 368. 22 La misma establece que la magnitud de una verificación interna a ser realizada localmente, en un laboratorio, depende de cuan completamente el método haya sido validado externamente. En base a eso, la norma clasifica los métodos de análisis en 6 categorías, dependiendo del grado de validación que se haya documentado.
En este caso concreto, al tratarse de un método previamente estandarizado como es el US EPA 8100, el alcance de la verificación interna que corresponde consiste en comprobar la veracidad, la precisión y los límites de cuantificación.
Para evaluar los parámetros de calidad del método durante la verificación interna se procedió de la siguiente forma:
Linealidad: se prepararon disoluciones a seis niveles de concentración dentro del intervalo de trabajo del método y se realizaron cuatro réplicas para cada caso. Cada disolución se inyectó por duplicado. Para cada analito, el intervalo de concentraciones de la curva de calibración se seleccionó de manera que el nivel de concentración más bajo fuera superior a su límite de cuantificación, mientras que el nivel más alto lo determinó la concentración de HAPs en la disolución patrón utilizada. Ver tabla 1.
Para evaluar la linealidad se realizó un análisis de varianza (ANOVA) en base a verificar la falta de ajuste del modelo. El ajuste al modelo lineal se probó con un nivel de significación del 95 % mediante la comparación del estadígrafo calculado F con Fα (k-2, n-k), donde k es el número de niveles de concentración (6) y n es el número total de observaciones. Si F calculado es menor que F0, 05(4, 22) = 3,88, la variabilidad debida a la falta de ajuste no es significativa y el modelo lineal explica la variabilidad de los datos.
Tabla 1 Niveles de concentración elegidos para el estudio de linealidad
| Compuestos | Niveles de concentración (µg/g) | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | ||
| Naftaleno | 0,09 | 0,43 | 1,10 | 2,20 | 5,28 | 9,80 | |
| Acenaftileno | 0,12 | 0,48 | 0,91 | 2,19 | 4,73 | 10,09 | |
| Acenafteno | 0,09 | 0,44 | 0,89 | 1,78 | 466 | 9,84 | |
| Fluoreno | 0,13 | 0,39 | 0,89 | 2,23 | 4,85 | 10,02 | |
| Fenantreno | 0,13 | 0,42 | 0,81 | 2,20 | 4,86 | 10,01 | |
| Antraceno | 0,12 | 0,36 | 0,83 | 2,12 | 4,83 | 10,03 | |
| Fluoranteno | 0,11 | 0,38 | 0,88 | 2,21 | 4,84 | 10,04 | |
| Pireno | 0,12 | 0,45 | 0,85 | 2,20 | 4,67 | 10,11 | |
| Benzo(a) Antraceno | 0,12 | 0,45 | 0,86 | 2,24 | 5,19 | 9,88 | |
| Criseno | 0,09 | 0,42 | 0,93 | 2,25 | 5,24 | 9,85 | |
| Benzo(b) Fluoranteno | 0,22 | 0,45 | 0,87 | 2,21 | 4,99 | 9,92 | |
| Benzo(k) Fluoranteno | 0,09 | 0,48 | 0,95 | 2,05 | 4,96 | 996 | |
| Benzo(a) Pireno | 0,09 | 0,46 | 0,85 | 2,17 | 5,32 | 9,75 | |
| Indeno(1,2,3-cd) Pireno | 0,14 | 0,43 | 0,80 | 2,20 | 4,80 | 10,03 | |
| Benzo(g,h,i) Perileno | 0,11 | 0,42 | 0,91 | 2,15 | 4,85 | 9,98 | |
Exactitud o veracidad: para la evaluación de este parámetro se empleó un sedimento de referencia certificado, que contenía a los 15 HAPs en estudio, a concentraciones dentro del intervalo de trabajo seleccionado para cada uno /11/. Se procesaron 8 réplicas empleando la extracción asistida con ultrasonidos y se calculó la recuperación según:
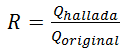 (2)
(2)donde R es la recuperación Q hallada la cantidad del analito recuperado después de procesar la muestra y Q original es el valor certificado de concentración.
Para evaluar las recuperaciones se utilizó, como criterio de comparación, el intervalo de 75-120 %, el cual se considera como aceptable para el caso de métodos cromatográficos.23 Igualmente, para cada caso se calculó el coeficiente de variación o RSD %, que se considera como aceptable cuando el valor calculado es inferior al 16 %.22
Precisión: se analizaron los parámetros repetibilidad y precisión intermedia. Para el estudio de repetibilidad se procesaron 8 réplicas del sedimento de referencia utilizado en el apartado anterior.18 Las extracciones asistidas con ultrasonidos fueron realizadas todas a la vez, por el mismo analista y en el mismo día. En paralelo se procesó un blanco bajo las mismas condiciones. Para la precisión intermedia se procesaron 10 réplicas del mismo sedimento. Las extracciones fueron realizadas por 3 analistas en días diferentes.
El desempeño de la repetibilidad y la precisión intermedia fue evaluado a partir de los resultados del coeficiente de variación para cada compuesto.
Límite de detección y cuantificación: Los límites de detección (LD) y cuantificación (LC) se obtuvieron a partir de la medición de un mínimo de 10 réplicas de un sedimento, tomado de una zona no contaminada con HAPs o que no ha sufrido impactos directos de este tipo de contaminantes. En este caso se realizó el pretratamiento anteriormente descrito, se procedió a la extracción mediante baño ultrasónico y a su posterior determinación por GC-FID. El cálculo de los límites de detección y cuantificación para cada compuesto fue realizado según lo propuesto en 23, específicamente para métodos cromatográficos.
Límites de detección (LD):
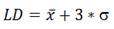 (3)
(3)
Límites de cuantificación (LC):
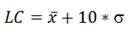 (4)
(4)
donde x es el valor medio de las respuestas del blanco y (, su Desviación Estándar.
Resultados y discusión
La inyección en el Cromatógrafo de gases de una disolución de referencia que contenía a los 15 HAPs en estudio, analizados bajo las condiciones operacionales establecidas en el método estandarizado US EPA 8100, dio como resultado el cromatograma que se muestra en la figura 1. De manera general se alcanza una adecuada resolución de los picos cromatográficos, excepto para los pares de isómeros, Fenantreno-Antraceno y Benzo(b)Fluoranteno-Benzo(k)Fluoranteno. Es conocido que la separación de isómeros por cromatografía de gases suele ser un problema de gran complejidad, no obstante, como se muestra en la figura ampliada, con el nivel de resolución alcanzado es posible determinar el área correspondiente a cada señal, con vistas a la determinación cuantitativa de cada uno de los compuestos.
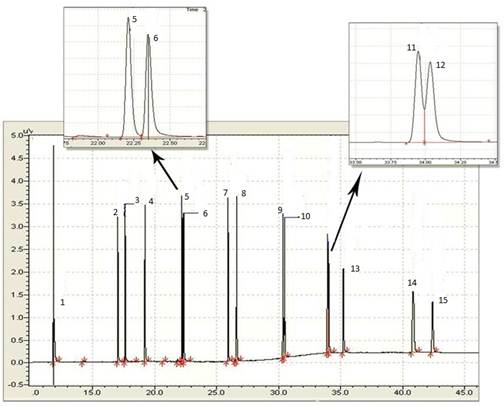
Fig. 1 Cromatograma correspondiente a la mezcla de los 15 HAPs en estudio. (1) Naftaleno, (2) Acenaftileno, (3) Acenafteno, (4) Fluoreno, (5) Fenantreno, (6) Antraceno, (7) Fluoranteno, (8) Pireno, (9) Benzo(a) Antraceno, (10) Criseno, (11) Benzo(b) Fluoranteno, (12) Benzo(k) Fluoranteno, (13) Benzo(a) Pireno, (14) Indeno(1,2,3-cd) Pireno y (15) Benzo(g,h,i) Perileno.
Verificación del método
Comprobación de la linealidad
Teniendo en consideración las concentraciones esperadas de los analitos de interés en las muestras y los valores de la recuperación para cada uno, se estableció el intervalo de concentraciones para estudiar el comportamiento de la linealidad en cada caso. En la tabla 2 se muestra este intervalo para cada compuesto en estudio.
Tabla 2 Intervalo de concentraciones para el estudio de linealidad
| Analitos | Concentraciones (µg/L) | |
|---|---|---|
| Límite inferior | Límite superior | |
| Naftaleno | 0,09 | 9,98 |
| Acenaftileno | 0,12 | 10,09 |
| Acenafteno | 0,09 | 9,84 |
| Fluoreno | 0,13 | 10,02 |
| Antraceno | 0,13 | 10,01 |
| Fenantreno | 0,12 | 10,03 |
| Fluoranteno | 0,11 | 10,04 |
| Pireno | 0,12 | 10,11 |
| Benzo(a)Antraceno | 0,12 | 9,88 |
| Criseno | 0,09 | 9,85 |
| Benzo(b)Fluoranteno | 0,10 | 9,92 |
| Benzo(k)Fluoranteno | 0,09 | 9,96 |
| Benzo(a)Pireno | 0,09 | 9,75 |
| IndenoPireno | 0,14 | 10,03 |
| Benzo(ghi)Perileno | 0,11 | 9,98 |
En la tabla 3, se muestran los resultados obtenidos del análisis de regresión, así como los principales estadígrafos obtenidos a partir de la correlación de la señal analítica (unidades de área de los picos cromatográficos) con la concentración a cada nivel, dentro del intervalo de trabajo.
Tabla 3 Resultados del análisis de regresión.
| Análisis de regresión | ||||||
|---|---|---|---|---|---|---|
| Compuesto | a | b | Anova | r | R2 | |
| Fcrítico | F calculada | |||||
| Naftaleno | 14525 | 7503 | 3,48 | 2,439 7 | 0,998 | 0,996 |
| Acenaftileno | 20142 | 472,49 | 2,716 5 | 0,997 | 0,995 | |
| Acenafteno | 21009 | 2567 | 0,530 8 | 0,999 | 0,999 | |
| Fluoreno | 20723 | 922,02 | 2,824 0 | 0,998 | 0,997 | |
| Antraceno | 20536 | 598,56 | 2,857 7 | 0,998 | 0,996 | |
| Fenantreno | 20670 | 498,11 | 2,231 0 | 0,998 | 0,996 | |
| Fluoranteno | 21761 | 428,10 | 2,312 9 | 0,999 | 0,998 | |
| Pireno | 21178 | 868,33 | 2,865 2 | 0,998 | 0,996 | |
| Benzo(a)Antraceno | 19936 | 619,18 | 2,440 5 | 0,998 | 0,997 | |
| Criseno | 19936 | 1552,39 | 2,795 7 | 0,999 | 0,998 | |
| Benzo(b)Fluoranteno | 20635 | 309,85 | 1,514 5 | 0,999 | 0,997 | |
| Benzo(k)Fluoranteno | 19892 | 2193,94 | 2,530 4 | 0,998 | 0,997 | |
| Benzo(a)Pireno | 18275 | 1104,97 | 2,563 1 | 0,997 | 0,994 | |
| IndenoPireno | 33018 | -2068,8 | 2,290 7 | 0,997 | 0,993 | |
| Benzo(ghi)Perileno | 22905 | 8,49 | 1,801 4 | 0,997 | 0,993 | |
Un primer criterio cualitativo para evaluar la calidad del ajuste es observar visualmente la medida en que los datos se ajustan a un modelo, es decir, si en este caso los puntos tienden a ajustarse razonablemente bien a una línea recta, tal como se muestra en la figura 2, que a modo de ejemplo, representa la curva de calibración obtenida para el Antraceno.

Un criterio cuantitativo para evaluar la linealidad lo proporciona el coeficiente de correlación. En todos los casos, los valores de R2 superiores a 0,99 obtenidos indican que los modelos lineales ajustados explican el 99 % de la variabilidad. Esto expresa que la calidad de los ajustes es satisfactoria y que, por ello, para todos los analitos, la relación matemática entre señal analítica (área debajo de los picos de cada compuesto) y niveles de concentración de la curva de calibración, es descrita adecuadamente por una línea recta. El coeficiente de correlación igual o superior a 0,99, indica una relación lineal estadísticamente significativa entre las variables. Esto se corresponde con uno de los criterios de aceptación establecidos para el análisis de trazas y con uno de los requerimientos planteados en la norma TS 368: 2010.22
La prueba de falta de ajuste o de Tiley 22 está diseñada para determinar si el modelo seleccionado es adecuado para describir los datos observados, o si se debería utilizar un modelo más complicado. Los resultados de esta prueba para el conjunto de HAPs en estudio se muestran en la tabla 4.
Tabla 4 Resultados del análisis de regresión. Prueba de falta de ajuste
| Compuesto | MR-IAEA-417 | Cmedia | Recobmedio | S% recuperación | CV% | TCalc. | TCrítica | ||
|---|---|---|---|---|---|---|---|---|---|
| µg/g | %Recob | ||||||||
| Naftaleno | 0,150 | 0,145 | 96,33 | 7,216 | 7,49 | 1,437 | 3,49 | ||
| Acenaftileno | 0,042 | ND | - | - | - | - | |||
| Acenafteno | 0,180 | 0,169 | 93,89 | 6,492 | 6,92 | 2,662 | |||
| Fluoreno | 0,230 | 0,222 | 96,52 | 3,973 | 4,12 | 2,476 | |||
| Fenantreno | 3,900 | 3,893 | 99,82 | 4,513 | 4,52 | 0,110 | |||
| Antraceno | 0,630 | 0,576 | 91,50 | 10,410 | 9,38 | 2,308 | |||
| Fluoranteno | 7,700 | 7,892 | 102,49 | 2,826 | 2,76 | 2,490 | |||
| Pireno | 6,000 | 6,467 | 107,78 | 7,897 | 7,33 | 2,787 | |||
| Benzo(a) Antraceno | 3,200 | 3,225 | 100,77 | 4,336 | 4,30 | 0,504 | |||
| Criseno | 3,600 | 3,753 | 104,26 | 5,719 | 5,75 | 0,835 | |||
| Benzo(b) Fluoranteno | 4,100 | 3,956 | 96,49 | 5,595 | 5,79 | 1,775 | |||
| Benzo(k) Fluoranteno | 2,000 | 2,212 | 110,60 | 6,931 | 6,67 | 1,515 | |||
| Benzo(a) Pireno | 2,800 | 2,707 | 96,69 | 7,218 | 7,47 | 1,295 | |||
| Indeno(1,2,3-cd) Pireno | 2,700 | 2,915 | 107,96 | 8,289 | 7,68 | 2,716 | |||
| Benzo(g,h,i) Perileno | 2,300 | 2,246 | 97,65 | 4,619 | 4,73 | 1,439 | |||
En todos los casos el valor del estadígrafo calculado (T calculado ) fue inferior a 3,49 (valor crítico), por lo tanto, la variabilidad debida a la falta de ajuste no es significativa y el modelo lineal explicó la variabilidad de manera satisfactoria. El análisis de los datos experimentales no sugirió la necesidad de considerar modelos superiores al primer orden por lo que se concluye que la correlación es lineal es la adecuada.
Veracidad
La tabla 4 también contiene los valores de recuperación media obtenidos a partir de las 8 réplicas analizadas, así como los niveles de concentración certificados para cada uno de los analitos en el material de referencia. Un primer criterio de la fiabilidad de los resultados obtenidos en este ensayo se expresa en los altos valores de recuperación obtenidos, todos en el intervalo entre 85 y 110 %. Con esto se cumple el criterio de aceptación establecido para métodos cromatográficos, cuando se trabaja a concentraciones del orden de 10-6.23 Otro de los criterios evaluados en el ensayo de veracidad fue el coeficiente de variación. Como se aprecia en la Tabla 4, sexta columna, para todos los analitos el coeficiente calculado fue inferior al 16%, que es el valor de referencia establecido en la normativa vigente, cuando se trabaja a concentraciones del orden de 10-6.22
Como complemento al análisis de veracidad según la metodología de verificación interna propuesta por la normativa empleada, se realiza una prueba-t para determinar si hay diferencia significativa o no, entre los resultados reportados en el material de referencia certificado y aquéllos obtenidos por el método a verificar. Para esta prueba se plantea la prueba-t, donde sí: tcalculada<tcrítica, los resultados observados no presentarán diferencias significativas respecto al material de referencia y viceversa. Todos los valores obtenidos fueron inferiores al tabulado de 3,49 (tabla 4), lo cual indica que no hay diferencias significativas entre los resultados de concentración obtenidos por el método internamente verificado y los reportados para el material certificado.
Estudio de la precisión del método
Repetibilidad
Se evaluaron los coeficientes de variación de los resultados obtenidos para cada compuesto en el análisis de 10 réplicas de un material de referencia, en condiciones de repetibilidad. Como se observa en la tabla 5, los resultados cumplen con lo establecido en la normativa como criterio de variabilidad entre replicas, es decir, que el valor obtenido de CV sea menor al 11% cuando se trabaja con concentraciones del orden de 10-6, aunque valores inferiores a 21% pueden considerarse aceptables.22
Tabla 5 Resultados obtenidos del análisis de repetibilidad
| Compuesto | CMR-IAEA-417 | Cmedia | CV% | |
|---|---|---|---|---|
| (µg/g) | ||||
| Naftaleno | 0,150 | 0,142 | 4,91 | |
| Acenaftileno | 0,042 | 0,037 | 7,93 | |
| Acenafteno | 0,180 | 0,165 | 7,91 | |
| Fluoreno | 0,230 | 0,202 | 6,97 | |
| Fenantreno | 3,900 | 3,891 | 4,49 | |
| Antraceno | 0,630 | 0,577 | 11,3 | |
| Fluoranteno | 7,700 | 7,881 | 2,49 | |
| Pireno | 6,000 | 6,311 | 6,71 | |
| Benzo(a) Antraceno | 3,200 | 3,200 | 3,90 | |
| Criseno | 3,600 | 3,575 | 5,37 | |
| Benzo(b) Fluoranteno | 4,100 | 3,945 | 5,62 | |
| Benzo(k) Fluoranteno | 2,000 | 2,071 | 6,31 | |
| Benzo(a) Pireno | 2,800 | 2,704 | 7,38 | |
| Indeno(1,2,3-cd) Pireno | 2,700 | 2,827 | 5,47 | |
| Benzo(g,h,i) Perileno | 2,300 | 2,220 | 3,81 | |
Precisión Intermedia
Los resultados fueron obtenidos a partir del análisis de un sedimento certificado, realizado en días y por analistas diferentes. Se utilizó el coeficiente de variación como criterio de medición de la variabilidad de las determinaciones. La tabla 6 muestra que los valores calculados para el CV, en todos los casos, se encuentran por debajo del límite establecido para este parámetro en la norma cubana.22
Tabla 6 Resultados obtenidos del análisis de precisión intermedia
| Compuesto | Cmedias µg/g | SR | CV |
|---|---|---|---|
| Acenaftileno | 5,90 ± 0,24 | 0,51 | 8,6% |
| Acenafteno | 3,25 ± 0,42 | 0,14 | 4,4% |
| Fluoreno | 1,52 ± 0,19 | 0,12 | 7,6% |
| Fenantreno | 4,78 ± 0,22 | 0,51 | 10,6% |
| Antraceno | 2,18 ± 0,11 | 0,53 | 7,0% |
| Fluoranteno | 8,34 ± 0,40 | 0,90 | 10,8% |
| Benzo(a) Antraceno | 6,05 ± 0,23 | 0,53 | 8,7% |
| Criseno | 2,52 ± 0,10 | 0,22 | 8,9% |
| Benzo(b) Fluoranteno | 4,22 ± 0,15 | 0,33 | 7,8% |
| Benzo(a) Pireno | 8,11 ± 0,20 | 0,47 | 5,7% |
| Indeno(1,2,3-cd) Pireno | 1,31 ± 0,07 | 0,11 | 8,3% |
En base a los resultados anterior se concluye que el método es preciso bajo las condiciones establecidas por la norma cubana TS 368: 2010.22
Límites de detección y cuantificación
La tabla 7 muestra los límites de detección y cuantificación calculados para cada uno de los compuestos, expresados en µg/g.
Tabla 7 Límites de detección y cuantificación
| Compuesto | Punto 1 µg/g | LD (µg/10g) | LC (µg/10g) |
|---|---|---|---|
| Naftaleno | 0,11 | 0,007 | 0,024 |
| Acenaftileno | 0,09 | 0,007 | 0,024 |
| Acenafteno | 0,13 | 0,012 | 0,041 |
| Fluoreno | 0,18 | 0,008 | 0,027 |
| Fenantreno | 0,13 | 0,006 | 0,020 |
| Antraceno | 0,12 | 0,006 | 0,019 |
| Fluoranteno | 0,11 | 0,006 | 0,019 |
| Pireno | 0,12 | 0,010 | 0,034 |
| Benzo(a) Antraceno | 0,12 | 0,009 | 0,031 |
| Criseno | 0,09 | 0,007 | 0,025 |
| Benzo(b) Fluoranteno | 0,12 | 0,006 | 0,019 |
| Benzo(k) Fluoranteno | 0,09 | 0,006 | 0,020 |
| Benzo(a) Pireno | 0,09 | 0,010 | 0,033 |
| Indeno(1,2,3-cd) Pireno | 0,14 | 0,009 | 0,031 |
| Benzo(g,h,i) Perileno | 0,11 | 0,005 | 0,016 |
Estos límites son similares, sobre todo para el Naftaleno, Fluoreno y Pireno a los encontrados por Peña y colaboradores, cuando utilizaron la extracción con Soxhlet para el estudio de la contaminación por HAPs en sedimentos.24
Los límites de cuantificación encontrados respaldan la utilidad del método para el estudio de la contaminación en sedimentos, aun cuando los HAPs se encuentren a bajas concentraciones. Estos límites son inferiores a los niveles máximos permisibles (TEL) y al nivel de exposición permisible (PEL) de contaminantes en sedimentos marinos y dulceacuícolas.25 El TEL y el PEL establecen la concentración de contaminantes sobre la cual aparecen con frecuencia efectos biológicos adversos.
Estudio de la contaminación por Hidrocarburos Aromáticos Policíclicos (HAPs) en muestras reales de sedimentos
Una vez verificado el método, se procedió a su aplicación en el análisis de muestras reales de sedimentos, tomadas en el embalse Hanabanilla, Villa Clara y en la bahía de Cienfuegos, para determinar la posible contaminación por HAPs.
Embalse Hanabanilla
Se estudiaron 15 HAPs considerados como prioritarios por la Agencia de Protección Ambiental de Estados Unidos (US EPA, por sus siglas en inglés) en muestras tomadas en un core de sedimentos. En la fracción superior del core (los tres primeros segmentos de 2 cm) que se corresponde con los sedimentos superficiales, fue posible cuantificar Antraceno, Fenantreno y Criseno según se muestra en la tabla 8. También se logró determinar el contenido como índice de HAPs totales que fue de 1,110 µg/g.
Tabla 8 Concentración de HAPs en sedimentos superficiales del Embalse Hanabanilla
| Compuesto | Conc.(µg/g) | UExp |
|---|---|---|
| Antraceno | 0,640 | ± 0,094 |
| Fenantreno | 0,330 | ± 0,028 |
| Criseno | 0,156 | ± 0,016 |
Es conocido el hecho de que las diferentes fuentes de contaminación generan una distribución característica de los HAPs, lo cual permite determinar el posible origen de la misma. Entre las relaciones diagnósticas más estudiadas para la contaminación por HAPs está la del fenantreno/antraceno.1 Cuando dicha relación es menor que 10, como en el presente caso (0,516), se considera que la contaminación fue originada por la acumulación de productos procedentes de procesos de combustión incompleta de materia orgánica, lo que indica que la contaminación encontrada en los sedimentos del embalse Hanabanilla tiene un origen pirogénico.
Bahía de Cienfuegos
Se tomaron muestras de sedimentos con el objetivo de estudiar el contenido de HAPs en las desembocaduras de los ríos Salado y Damují hacia la bahía de Cienfuegos Estos se encuentran ubicados en el lóbulo norte de la bahía o zona industrial 2, como se observa en la figura 3. Las muestras se corresponden con el período seco del año 2014. Los sitios de muestreo están ubicados en las estaciones definidas para evaluar los aportes de acuatorios hacia la bahía, según lo establecido en el programa nacional de monitoreo a bahías. La localización de las estaciones de muestreo está relacionada con los impactos probables desde fuentes de contaminación al ecosistema primario, ya sea ríos, vertimientos directos o indirectos desde fuentes en tierra y las mismas coinciden con las estaciones del muestreo hidrológico, biológico y de sedimentos de la bahía.

En el sedimento correspondiente a la estación del Río Salado fue posible determinar seis de los HAPs en estudio (tabla 9), a concentraciones similares a las encontradas por Tolosa y colaboradores en 2009 16 en cuanto a los niveles de Fluoreno, Antraceno y Pireno. En el caso del Fenantreno y el Antraceno, las concentraciones actuales fueron inferiores a las descritas en aquel estudio.
Para el caso de la estación ubicada en la desembocadura del Río Damují, los resultados difieren de los obtenidos en ese estudio previo, encontrándose ahora concentraciones más bajas de los HAPs presentes (tabla 9). La diferencia pudiera estar asociada, entre otras causas, a una atenuación o disminución de la fuente emisora de estos compuestos.
Es importante señalar que en las desembocaduras de ambos ríos, las concentraciones de todos los HAPs cuantificados supera el valor TEL 25 y que en ambas estaciones de monitoreo, se cuantificaron niveles de HAPs superiores al umbral PEL para Fluoreno, Antraceno y Pireno, lo cual tiene un impacto negativo directo sobre la salud de los organismos de la zona béntica.

Conclusiones
La adecuación y verificación interna del método US EPA 8100 permitió disponer de un procedimiento basado en la extracción asistida por ultrasonidos y determinación por CG, para la determinación fiable en muestras de sedimentos de 15 HAPs reconocidos como contaminantes prioritarios. El método verificado resultó ser preciso, exacto y lineal en los intervalos de concentración estudiados para cada compuesto. Los límites de detección y cuantificación encontrados son similares a los obtenidos a nivel internacional para métodos equivalentes, empleados en la evaluación de estos contaminantes en muestras de sedimentos. La aplicación del método a muestras reales permitió demostrar la contaminación por HAPs en los sedimentos del embalse Hanabanilla y en las desembocaduras de los ríos Salado y Damují en la bahía de Cienfuegos

