










 Custom services
Custom services Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La mucopolisacaridosis tipo I (MPS-1) es una enfermedad metabólica lisosomal debida a un déficit enzimático de alfa-L-iduronidasa, que se traduce en una acumulación de glucosaminoglucanos (GAG) dermatán y heparán sulfato en órganos y tejidos y en el aumento de su excreción urinaria. Es una enfermedad poco frecuente, con una incidencia de aproximadamente uno de cada 100 000 recién nacidos vivos. Se hereda de forma autosómica recesiva y hay descritas más de 80 mutaciones diferentes del gen IDUA (locus 4p16.3).1 Fue descrita en 1919 por Gertrud Hurler, que publicó la historia clínica de pacientes con opacidad corneal y retardo mental. En 1952 Brante aisló el mucopolisacárido dermatán sulfato en el hígado de dos pacientes con síndrome de Hurler; la enfermedad recibió el nombre de mucopolisacaridosis (MPS). Las otras dos formas de la enfermedad son el síndrome de Sheie, que es la forma más leve de MPS y el síndrome de Hurler Sheie, que es menos severo. El síndrome de Hurler es el más grave de los subtipos de MPS-1, el niño presenta retraso en el neurodesarrollo antes del primer año de vida y se detiene entre las edades de dos a cuatro años. A esto le siguen un deterioro y la pérdida progresiva de las capacidades mentales y físicas. El lenguaje puede sufrir limitaciones debido a la pérdida de la audición y a la macroglosia; también presentan opacidad corneal y síndrome de túnel carpiano. Los niños afectados pueden ser muy grandes al nacer, pero al año comienza a retrasarse la estatura y se detiene a los tres años de edad, presentan un tronco corto, las características faciales incluyen frente prominente, puente nasal plano, cejas muy pobladas, boca abierta y paladar ojival.2 En algunos casos son frecuentes la miocardiopatía y las anomalías valvulares, la organomegalia, las hernias y el hirsutismo. El diagnóstico temprano es difícil, se basa en la determinación del déficit de la enzima alfa-L-iduronidasa, la detección de un incremento de heparán y dermatán sulfato en la excreción urinaria y la prueba genética confirmatoria de la mutación del gen IDUA.3 El pronóstico es desalentador, los niños con esta enfermedad presentan problemas del sistema nervioso y pueden morir a temprana edad por complicaciones respiratorias y cardíacas.
INFORMACIÓN DEL PACIENTE
Se presenta un adolescente masculino de 12 años de edad, con antecedentes de ser producto de un embarazo bajo riesgo obstétrico, parto eutócico a las 39,5 semanas, institucional, con peso al nacer de 7,10 libras, que presentó hipocalcemia neonatal, sin otras complicaciones.
Caminó al año y comenzó el círculo infantil, a los dos años la educadora se percata de que dejó de hablar y tenía babeo constante. Fue llevado a la consulta de un Especialista en Neurología del Hospital Pediátrico Universitario “José Luis Miranda” de la Ciudad de Santa Clara, Provincia de Villa Clara, que solicitó una interconsulta con el Centro de Diagnóstico y Orientación y le fue diagnosticado un posible retraso mental severo. El Especialista en Neurología indicó como tratamiento levomepromazina, ½ tableta cada 12 horas, carbamazepina, una tableta cada ocho horas y vitamina B6, tres tabletas al día.
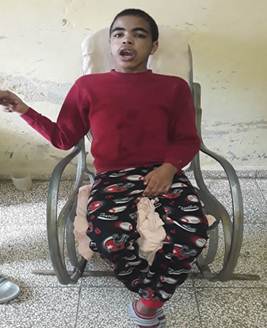
Los Especialistas en Neurología y Genética, por sus características fenotípicas y su retardo en el neurodesarrollo, sospecharon un posible síndrome de Hurler y le indicaron estudios correspondientes. Al culminar pre-escolar le diagnosticaron déficit de atención y fue trasladado al Hogar de impedidos físicos, en el que estuvo hasta los nueve años; la mamá decidió dejarlo en el hogar cuando dejó de caminar. Al examen físico se constataron fascie tosca, cara alargada, frente ancha, cejas muy pobladas, labios gruesos, paladar ojival, tendencia a la boca abierta, babeo constante, puente nasal deprimido e hipertricosis en la espalda (Figuras 1 y 2). En el sistema osteomioarticular: escoliosis toracolumbar, deformidad de las rodillas y rigidez articular, en el sistema genitourinario: macroorquidismo, a la palpación abdominal: hepatomegalia y esplenomegalia, el percentil talla/edad: 90-97 y el fondo de ojo normal.
Estudios realizados:
Potenciales evocados de tallo y tronco cerebral: hipoacusia ligera en oído derecho y moderada en oído izquierdo
Electroencefalograma: normal
Tomografía axial computadorizada (TAC) de cráneo: atrofia cortical
Ultrasonido abdominal: hepatomegalia de 5 cm y esplenomegalia de 2 cm
Ecocardiograma normal
Cromatografía de mucopolisacáridos: presencia de dermatán y heparán sulfato
Hemograma y química sanguínea dentro de límites normales.
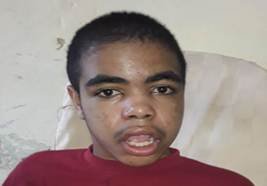
Figura 1 Muestra fascie tosca, frente ancha, cejas muy pobladas, labios gruesos y puente nasal ancho
El diagnóstico se confirmó por estudio de cromatografía de mucopolisacáridos realizado en el Centro Nacional de Genética.
DISCUSIÓN
Esta afección es una enfermedad metabólica congénita. Un desorden de carácter autosómico recesivo que resulta de mutaciones en el gen codificador de la enzima alfa-L-iduronidasa, por lo que se produce una acumulación de los GAGs heparán sulfato y dermatán sulfato en todos los tejidos del organismo y causan una amplia variedad de síntomas físicos, así como anormalidades que dañan los órganos, entre ellos el corazón. Estas moléculas de mucopolisacáridos se encuentran en todo el cuerpo, a menudo en las secreciones mucosas y en el líquido que rodea las articulaciones. La tasa de acumulación es variable en cada persona afectada y esto hace que haya una gran diversidad en las manifestaciones clínicas; los síntomas se hacen más aparentes a medida que avanza la acumulación. Ambos padres necesitan trasmitir el gen defectuoso para que su hijo desarrolle este síndrome.4
Dentro de los diagnósticos diferenciales de esta enfermedad se encuentran:
Síndrome Hurler-Scheie: representa el fenotipo intermedio de una deficiencia de alfa-L-iduronidasa. Estos pacientes presentan talla corta, problemas dentales y alto riesgo de complicaciones asociadas con anestesia. Compromiso severo de las articulaciones e infecciones respiratorias y otitis crónicas, tórax pequeño, hepatoesplenomegalia, rasgos faciales toscos y opacidad corneal, hernia umbilical e inguinal, fascies dismórfica, retraso del desarrollo, hipoacusia, problemas gastrointestinales, deformidades esqueléticas y defectos cardíacos (causados por acumulación de dermatán sulfato), entre otros; sin embargo, su sistema nervioso es normal.5
Síndrome de Scheie: es la forma más leve de una deficiencia de alfa-L-iduronidasa, se presenta con mayor frecuencia en los niños a partir de los cinco años de edad. A diferencia de los demás tipos este tiene un índice de mayor incidencia en presentar glaucoma. Estos pacientes, por lo general, tienen una estatura normal y su hepatoesplenomegalia es muy leve. La rigidez articular puede no ser evidente, no presentan retardo y no tienen rasgos toscos. La disostosis múltiple es muy leve. El síndrome de túnel del carpo u otra manifestación de rigidez articular es lo que hace que estos pacientes se presenten al médico; los problemas cardíacos se ven asociados a la acumulación de mucopolisacáridos en las venas y las arterias. El deterioro ocular, como la opacidad corneal y la degeneración de la retina, puede llevar a la sospecha de una mucopolisacaridosis.5
Síndrome de Hunter: es determinado por una mutación en el cromosoma X, en la región Xq25-q27, que afecta la función normal de la enzima iduronato sulfatasa, esencial para la fragmentación de dos mucopolisacáridos, dermatansulfato y heratansulfato, que provoca la acumulación citoplasmática de mucopolisacáridos. Las características clínicas se instalan progresivamente. Consisten en fascie tosca, piel engrosada, hernia umbilical, hepatoesplenomegalia, giba, base nasal ancha, baja talla y rigidez articular.6
En cuanto al diagnóstico de la enfermedad es posible realizarlo en la etapa prenatal al medir la actividad enzimática en un cultivo de vellosidades coriónicas o de amniocitos, a través de una prueba genética si se conoce la mutación responsable de la enfermedad. Otro método se basa en la detección de un incremento de heparán y dermatán sulfato en la excreción urinaria y se confirma demostrando la deficiencia enzimática de alfa-L-iduronidasa.3
El diagnóstico definitivo se establece por el estudio genético molecular del tipo de mutación del gen IDUA,1 no disponible en Cuba. Se hereda como un rasgo genético autosómico recesivo, la alteración cromosómica ha sido identificada y se localiza en el brazo corto del cromosoma 4, cercano al gen que regula la expresión de la enfermedad de Huntington. Se han descrito numerosas mutaciones con diferentes grados de expresión, lo que explica la gran variabilidad de formas clínicas en esta enfermedad.7 Otro estudio se basa en la demostración de la actividad deficiente de la enzima lisosomal alfa-L-iduronidasa en los leucocitos de sangre periférica, los fibroblastos cultivados o de plasma. La excreción urinaria de glucosaminoglucano (heparán y sulfato de dermatán) es una prueba preliminar útil.8
Para determinar la afectación de otros sistemas se deben realizar estudios tales como: potenciales evocados de tronco y tallo cerebral, ecocardiograma, electroencefalograma y ultrasonido abdominal, entre otros, según se estime.
El manejo de la enfermedad es multidisciplinario. El trasplante de precursores hematopoyéticos es el tratamiento de elección para pacientes con síndrome de Hurler menores de dos años y medio de edad porque puede prolongar la supervivencia, preservar la neurocognición y mejorar algunas características somáticas. La terapia enzimática sustitutiva con alfa-L-iduronidasa se recomienda para todos los pacientes y es una terapia de por vida que alivia los síntomas no neurológicos. El manejo adicional de esta enfermedad es, en gran medida, de apoyo e incluye intervenciones quirúrgicas (adenoamigdalectomía, reparación de hernias, derivación ventriculoperitoneal y remplazo de válvula cardíaca) y la medicación para aliviar los síntomas gastrointestinales.3
El síndrome de Hurler es la forma más grave de las mucopolisacaridosis. Su evolución progresiva lleva, por lo general, a la muerte antes de los 10 años por causas cardiorespiratorias,1,2,3,8 pero el trasplante de precursores hematopoyéticos puede mejorar la esperanza de vida, así como la terapia enzimática con alfa-L-iduronidasa.3
Las MPS son un amplio grupo de enfermedades infrecuentes, pero con un impacto para el paciente, la familia y la sociedad muy alto. Es importante reconocer sus características para poder establecer un diagnóstico oportuno y ofrecer un tratamiento adecuado.9
Las pruebas y el asesoramiento genético, al igual que tener un perfil completo de los antecedentes familiares, son importantes para los futuros padres con historia familiar del síndrome de Hurler.
Se realizó la presentación de un caso interesante, no comúnmente visto en la práctica médica, con el objetivo de dar a conocer a estudiantes y profesionales de la salud las características físicas del paciente con este síndrome, el que ha tenido una supervivencia mayor de los 10 años de vida, gracias a la atención y al manejo multidisciplinario.

