










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La enfermedad granulomatosa crónica (EGC) es una inmunodeficiencia primaria (IDP) congénita del sistema inmune innato, originada por defectos en el complejo enzimático nicotinamida adenina dinucleótido fosfato (NADPH) oxidasa o sistema NADPH oxidasa, presente en células fagocíticas, tales como: macrófagos, neutrófilos, monocitos y eosinófilos. Estos defectos funcionales causan deficiencia en la producción de especies reactivas del oxígeno (EROs) de los fagocitos, afectando la necesaria eliminación de algunos microorganismos patógenos dentro del fagolisosoma.1,2,3 La enfermedad se manifiesta con mayor frecuencia en la infancia con prevalencia de 1 caso por cada 200 000 o 250 000 nacidos vivos, sin preferencia étnica.3,4,5 En el mundo occidental, 70 % de los pacientes muestran un patrón de herencia genética ligada al cromosoma X, en los que la descendencia afectada dependerá del sexo del progenitor que porta la mutación; mientras que el resto muestra un patrón de herencia de tipo autosómica recesiva, en la que ambos padres son portadores de la mutación en una de sus dos variantes alélicas autosómicas.6 En países con elevada tasa de matrimonios consanguíneos, la EGC autosómica recesiva suele ser más común que la ligada al cromosoma X.2 Por otro lado, los pacientes con EGC por herencia ligada al X son el doble de susceptibles a sufrir complicaciones inflamatorias con respecto a los pacientes con herencia autosómica recesiva de la enfermedad.1
La EGC se identificó por primera vez en la década de 1950 en un niño norteamericano de 12 meses que presentaba linfadenitis crónica supurativa, hepatoesplenomegalia, infiltrados pulmonares y dermatitis eczematoide. En la descripción clásica, de 1959, de la “enfermedad granulomatosa mortal de la infancia”, Bridges y otros, informaron: “no se ha encontrado una terapia satisfactoria, y la enfermedad ha progresado implacablemente a través de un debilitamiento severo hasta la muerte final en un período de varios años” 7) En virtud de los avances científicos, desde 1959 hasta la actualidad, la supervivencia de los pacientes con EGC ha ido en aumento, gracias a la combinación de profilaxis antibacteriana, antifúngica e inmunomoduladora de por vida, el trasplante de médula ósea hematopoyético y la terapia génica.1,8,9,10,11 El punto de partida decisivo para lograr todo lo expuesto anteriormente es el diagnóstico certero de la enfermedad, pues de este modo el paciente podrá recibir el tratamiento personalizado que requiera, y por tanto, elevar su esperanza y calidad de vida.
El objetivo de este trabajo es describir los aspectos fisiopatológicos y moleculares de la EGC; así como, discutir aspectos relacionados con dos de las pruebas empleadas actualmente para lograr su diagnóstico: la prueba de reducción de nitroazul de tetrazolio (NBT) y la de 1,2,3-dihidrorodamina (DHR).
Métodos
Se llevó a cabo una investigación bibliográfica-documental a partir de artículos científicos publicados entre los años 1933 y 2018, para ello fueron consultadas las bases de datos SciELO, PubMed y Springer.
Análisis y síntesis de la información
NADPH oxidasa
El sistema NADPH oxidasa (también conocido como NOX2) comprende 5 glicoproteínas y una proteína G; el heterodímero unido a la membrana: p22phox y Nox2 o 91phox, que constituye el centro catalítico (se encuentran asociadas en la membrana formando un heterodímero, tanto en la forma activa del complejo como en la inactiva) y 4 subunidades que se localizan en el citosol en su estado basal: p47phox (NCF1), p67phox (NCF2), p40phox (NCF4) y la proteína G denominada Rac (GTPasa Rac), que se encuentra unida a GDP y a Rho-GDI en su forma inactiva (Fig. 1). Durante el proceso de activación de este complejo, las proteínas citosólicas fosforiladas por acción de la proteína quinasa C, se traslocan a la porción unida a la membrana y ensamblan el complejo oxidasa funcional de 6 componentes.12,13 El complejo NADPH oxidasa también depende de la asociación de las GTPasas citosólicas de bajo peso molecular: Rac1 y Rac2, las cuales permiten la activación del complejo y están involucradas en la regulación del sistema NADPH oxidasa. 6,14) (Fig. 1) La NADPH oxidasa activada es capaz de efectuar la transferencia de electrones de NADPH a FAD y al grupo hemo, para reducir el oxígeno molecular (O2) a anión superóxido (O2 -) microbicida y otros agentes citotóxicos, como el peróxido de hidrógeno, que son responsables de la oxidación de proteínas, lípidos, carbohidratos y ácidos nucleicos presentes en agentes infecciosos mediante el proceso denominado estallido respiratorio.3,6
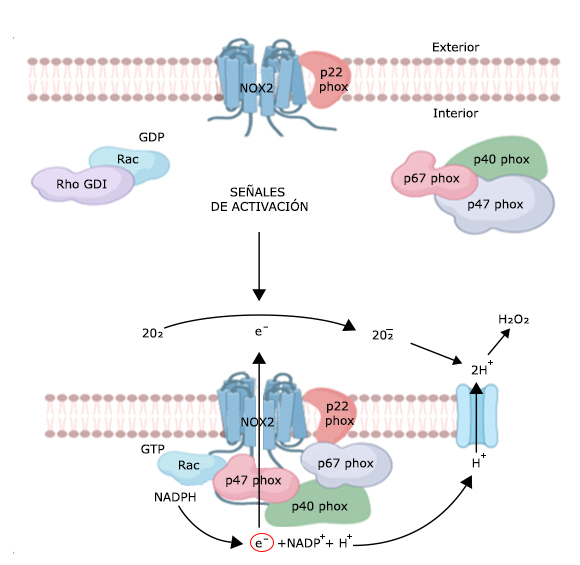
Fig. 1 Representación esquemática de los mecanismos moleculares del complejo de activación NADPH oxidasa (NOX 2) en la membrana citoplasmática de un gránulo de neutrófilo.
El término “estallido respiratorio” fue acuñado inicialmente por Baldridge y Gerrard en 1933,15 después de descubrir que los neutrófilos consumen grandes cantidades de oxígeno durante la fagocitosis. El mecanismo del estallido respiratorio no está relacionado con la vía de fosforilación oxidativa mitocondrial, pues se comprobó que inhibidores mitocondriales clásicos como el cianuro no afectaban el proceso;8,16 pero se conoce que la capacidad de los neutrófilos de destruir bacterias ya internalizadas se daña en ausencia de oxígeno.8 El componente molecular responsable del estallido respiratorio es un flavocitocromo con un máximo de absorbancia cercano a 558 nm que se encuentra unido a la membrana, al que se denominó “flavocitocromo b558”.8,17 El flavocitocromo b558 es el mismo heterodímero antes mencionado compuesto por p22phox y Nox2.8 (Fig. 1) Aproximadamente el 85 % del flavocitocromo b558 se expresa constitutivamente en las membranas de los fagocitos, y un porcentaje menor se localiza en el plasma celular y las membranas secretoras de los gránulos.8,18
Fisiopatología
Un avance importante en el estudio de la fisiopatología de esta enfermedad se produjo cuando Quie y otros hallaron que la fagocitosis y la muerte intracelular no están vinculadas en los pacientes con EGC;19 pues los fagocitos pueden moverse e internalizar microorganismos de forma normal; pero no pueden destruir determinados hongos y bacterias denominados catalasa positivos, debido a su incapacidad de metabolizar el oxígeno para producir EROs.2,3,6 Los organismos catalasa positivos (por ejemplo, Staphylococcus aureus, Pseudomonas, Aspergillus fumigatus, Paecilomyces variotti, Burkholderia cepacian, Salmonella, Nocardia, Candida albicans, Enterobacteriaceae, Klebsiella spp y Serratia spp) pueden descomponer el peróxido de hidrógeno producido por el huésped en oxígeno y agua, mediante la enzima catalasa.20,21 En humanos, el peróxido de hidrógeno generado por el sistema NADPH oxidasa, también sirve de sustrato a enzimas como las mieloperoxidasas (MPO) para la formación en presencia de cloro, del ácido hipocloroso, otro potente agente oxidante contra patógenos.22
Además de tener efectos citotóxicos directos, la producción de EROs tiene influencia en otras funciones de la inmunidad innata. Por ejemplo, los pacientes con EGC muestran una expresión y función reducidas de los receptores tipo Toll, los receptores del complemento y los receptores de quimiocinas, que se correlacionan con la gravedad de la enfermedad.23 Además, los neutrófilos también utilizan medios extracelulares de actividad microbicida mediante el uso de trampas extracelulares de neutrófilos.24 Este mecanismo depende de la producción normal de EROs por el sistema NADPH oxidasa.25Marzaioli y otros encontraron que la diferenciación y maduración de células dendríticas y monocitos también se ven afectadas por alteraciones en el sistema NADPH oxidasa.26
Las manifestaciones clínicas más frecuentes son: linfadenopatía, hepatoesplenomegalia, neumonía recurrente; además de otras manifestaciones como conjuntivitis, sinusitis, dermatitis, estomatitis ulcerativas, diarrea crónica, obstrucción intestinal y osteomielitis supurativa.3,6,27 El primer hallazgo clínico suele ocurrir en edades tempranas y suele ser una adenitis en el área de aplicación de la vacuna BCG, pues algunos pacientes con EGC tienen predisposición a la infección por Mycobacterium tuberculosis.2,28 También son comunes los procesos inflamatorios por desregulación del sistema inmunitario y la autoinmunidad. 1
Los fagocitos al no lograr controlar las infecciones, estimulan respuestas inmunitarias celulares crónicas, lo que da lugar a la activación de macrófagos mediada por linfocitos T y a la formación de granulomas compuestos de macrófagos activados, los cuales intentarán eliminar los patógenos a pesar de la producción defectuosa de EROs. La formación de granulomas es un evento inflamatorio clásico en pacientes con EGC, en tal medida, que este aspecto histológico constituye la base del nombre de la enfermedad. Distintos de los relacionados con otras enfermedades, los granulomas en la EGC generalmente no son caseificantes, se encuentran dentro de los tejidos fibróticos en el contexto de inflamación aguda o crónica.29 En pacientes con EGC, los granulomas son propensos a afectar las vísceras huecas, especialmente el colon, el estómago y la vejiga, en dichos granulomas se encuentran macrófagos activados que dan lugar a obstrucciones uretrales, pilóricas y esofágicas.30,31
Caracterización genética
Toda mutación patológica que tenga lugar en los genes que codifican las subunidades proteicas del sistema NADPH oxidasa, inducen la EGC (Fig. 2).2,4,8,32 Existe un amplio rango en la capacidad de generar EROs entre los pacientes de EGC, que generalmente varía desde indetectables (0 %) hasta menos del 30 %.33 Aquellos pacientes con mutaciones menos comprometedoras, tienen una producción residual de EROs y no presentan manisfestaciones clínicas graves.34
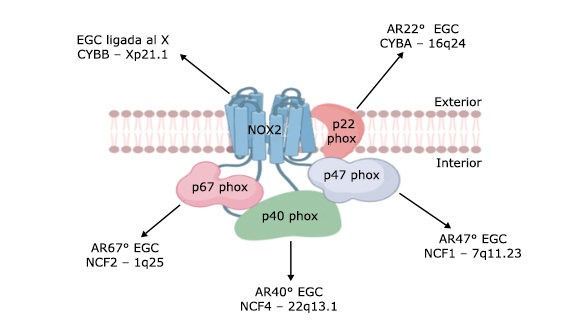
Fig. 2 Mutaciones patológicas del sistema NADPH oxidas (NOX 2) relacionadas con la enfermedad granulomatosa crónica (ECG).
El patrón de herencia genética más frecuente es la (EGC) ligada al cromosoma X. Otras formas genéticas existentes tienen un patrón de herencia autosómico recesivo (AR): AR-CGD47o, AR-CGD67o y AR-CGD22o.
La causa más común de EGC es un defecto en el gen CYBB (Nox2), ubicado en el brazo corto del cromosoma X (Xp21.1-p11.4). Múltiples tipos de mutaciones del gen CYBB (por ejemplo, deleción, cambio de marco de lectura, mutación sin sentido y mutaciones en el sitio de empalme) pueden provocar la no expresión o reducción de la expresión del producto génico. La EGC relacionada con CYBB es heredada de forma recesiva ligada al cromosoma X; sin embargo, se estima que del 10 % al 15 % de las mutaciones en Nox2 son resultado de nuevas mutaciones que ocurren en la línea germinal. (8)
Aunque la base genética de la EGC en casi dos tercios de los pacientes con la enfermedad radica en el locus CYBB, también se reportan mutaciones bialélicas (generalmente un codón de parada prematuro) en el factor citosólico 1 de neutrófilos (NCF1, 7q11.23), que da como resultado un daño en la función biológica del componente p47phox.8 La EGC relacionada con NCF1 se hereda de forma autosómica recesiva; en este caso, ambos padres serían portadores asintomáticos de una sola mutación NCF1.8
Los hombres con una mutación en CYBB o mutaciones bialélicas en CYBA o NCF1, NCF2 o NCF4, presentan manifestaciones clínicas de EGC. Por otro lado, las portadoras femeninas de una sola mutación en CYBB exhiben una actividad NADPH oxidasa afectada en una porción de sus fagocitos, más comúnmente del 20 % al 80 %, como resultado de la inactivación aleatoria del cromosoma X o fenómeno de Lyonización. 35 Este fenómeno fue descrito por la doctora Mery Lyon en 1961, y consiste en el silenciamiento al azar de un cromosoma X en todas las células somáticas del organismo femenino, con el objetivo de lograr compensación de la dosis génica.36 El estado de portador puede provocar una variedad de síntomas, que incluyen: úlceras aftosas, artralgias y fotosensibilidad cutánea.11 En un estudio de 162 portadores de EGC con herencia ligada al X. Marciano y otros, encontraron que las complicaciones infecciosas eran altamente probables en aquellos pacientes con menos del 10 % de actividad de la DHR,37 incluso los pacientes con 10 % al 20 % de actividad de DHR permanecieron en un riesgo sustancialmente mayor de infección. Por esta razón, se aconseja prudente administrar el tratamiento para esta enfermedad cuando la actividad de la DHR cae por debajo del 20 %.37
Pueden surgir complicaciones adicionales cuando la región del gen Xp21.1 (CYBB) posee una deleción génica más sustancial. Las deleciones lo suficientemente grandes como para abarcar tanto el gen CYBB como el locus Kell (gen XK) conducen a un síndrome de deleción génica contigua;38 en estos casos se debe considerar la EGC como diagnóstico, particularmente si hay síntomas clínicos relacionados con esta enfermedad.8
Existen reportes de mutación dominante negativa en Rac2 en dos infantes masculinos.39,40 El primero presentó infecciones en tejidos blandos, neutrofilia y deficiencia en la quimiotaxis de los neutrófilos.39,41 El segundo paciente presentaba disfunción neutrofílica, leucocitosis, linfopenia CD4+ y niveles reducidos de IgA e IgM.40
Pruebas de diagnóstico
En aquellos pacientes con antecedentes clínicos compatibles, se indica realizar el diagnóstico de EGC mediante la evaluación de la funcionalidad del complejo NADPH oxidasa en neutrófilos estimulados.42,43 La prueba del nitroazul de tetrazolio (NBT) se ha utilizado históricamente para medir la formación del anión superóxido. En esta prueba, los neutrófilos se estimulan con acetato de forbolmiristato (PMA) en presencia de colorante NBT.44 Después de la estimulación, el colorante amarillo es reducido por el complejo NADPH oxidasa en neutrófilos normales a formazán, que es un precipitado azul oscuro o púrpura que se retiene dentro de la célula. Las células luego se analizan visualmente por microscopía para detectar este cambio de coloración. La mayoría de los neutrófilos normales será de color azul oscuro, pero los neutrófilos que carecen de un complejo NADPH funcional no cambian de color.4
La prueba NBT es sencilla y económica, pero con limitaciones importantes que deben ser tomadas en consideración si el diagnóstico de un paciente dependerá enteramente de su resultado. Esta prueba a veces puede diagnosticar portadoras femeninas ligadas al cromosoma X, porque deberían tener una población mixta de células positivas y negativas como resultado del fenómeno de Lyonización. Sin embargo, la lectura manual de la prueba NBT es semicuantitativa y, muy a menudo las portadoras de mutaciones ligadas al cromosoma X no pueden ser bien identificadas. Los portadores de una mutación autosómica recesiva tampoco se distinguen bien en esta prueba, porque las células necesitan solo una pequeña cantidad de NADPH oxidasa funcional para reducir la NBT. Aunque el ensayo NBT requiere poca sangre para realizarse, lo ideal es que la sangre sea fresca, lo que significa que una muestra no sería útil si es necesario transportarla desde grandes distancias. Otros problemas adicionales que tiene este tipo de prueba son los diagnósticos falsos positivos o negativos entre las portadoras de EGC ligados al cromosoma X.45 Además, la confiabilidad de la prueba depende enteramente de la experiencia en el microscopio óptico del especialista a cargo de realizarla.4
Desde finales de la década de 1990, la prueba NBT ha sido reemplazada en gran medida por la prueba DHR por citometría de flujo como pilar fundamental para diagnosticar la EGC.46,47 En comparación con la prueba NBT, el ensayo DHR es más fácil de realizar, más confiable, más cuantitativo y más sensible. Similar a la prueba NBT, los fagocitos se estimulan con PMA o lipopolisacárido (LPS) bacteriano; pero luego se incuban con 1,2,3-dihidrorodamina, que penetra en las células libremente y es oxidada intracelularmente por peróxido de hidrógeno u otro EROs (producido en presencia de un sistema normal de NADPH oxidasa-MPO) a 1,2,3-rodamina, la cual emite una señal de 585 nm en el espectro fluorescente cuando es excitada por una luz con longitud de onda de 488 nm.48
El paso siguiente del proceso es evaluar la fluorescencia de las células estimuladas como una medida sustituta de la actividad de la NADPH oxidasa; mediante el citómetro de flujo se logra detectar la señal de fluorescencia emitida por cada célula por separado, de ahí la gran sensibilidad de este tipo de prueba.49 La intensidad de fluorescencia media de la 1,2,3-rodamina se correlaciona cuantitativamente con la producción intermedia de oxígeno reactivo y la supervivencia posterior en pacientes con EGC.34 Mediante esta prueba se puede diferenciar entre la EGC ligada al cromosoma X, la EGC autosómica recesiva y el estado de portador ligado al X en mujeres.46 También, se puede evaluar la actividad residual de la enzima, la medición del estallido respiratorio en neutrófilos y monocitos por separado, y el grado de Lyonización en las portadoras, que se ha correlacionado con un mayor riesgo de infección en mujeres que tienen una población normal de neutrófilos de menos del 20 % como se mencionó en el acápite anterior.4,37,49
Resulta importante mencionar que como esta reacción de oxidación es dependiente de la peroxidasa y por tanto, se basa en la actividad de la MPO o peroxidasa eosinofílica en los fagocitos; en caso de deficiencia de MPO, una condición no infrecuente, el ensayo DHR dará un resultado negativo, el cual puede ser malinterpretado como una deficiencia en el sistema NADPH oxidasa; o sea, como un paciente falso positivo a EGC.50
La EGC puede generar consecuencias inmunológicas e inflamatorias graves, conduciendo a infecciones potencialmente mortales, procesos inflamatorios por desregulación del sistema inmunitario y autoinmunidad. El diagnóstico de pacientes con síntomas y signos consistentes con dicha enfermedad, se puede realizar mediante la prueba DHR, que es una forma rápida y confiable de evaluar la actividad de la NADPH oxidasa, si se cuenta con los recursos necesarios para su realización. Una vez obtenido el diagnóstico, los resultados de la citometría de flujo de la DHR junto a la secuenciación genética, pueden ayudar a predecir la gravedad de la enfermedad. También, se recomienda realizar estos estudios a los familiares directos de aquellos pacientes positivos a la EGC; además, las pruebas genéticas y prenatales pueden ser útiles para la detección de pacientes portadores.

