










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMEDISAN
versión On-line ISSN 1029-3019
MEDISAN vol.16 no.3 Santiago de Cuba mar. 2012
ARTÍCULO DE REVISIÓN
Hemostasia normal y coagulación intravascular diseminada en obstetricia
Normal hemostasis and disseminated intravascular clotting in Obstetrics
Dr.C. Danilo Nápoles Méndez I y MsC. Dianela Nápoles García II
I Hospital Ginecoobstétrico Provincial Docente "Mariana Grajales Coello", Santiago de Cuba, Cuba.
II Hospital General Docente "Dr. Juan Bruno Zayas Alfonso", Santiago de Cuba, Cuba.
RESUMEN
En el período gravido - puerperal, la coagulación sanguínea sufre cambios que se impone conocer para interpretar correctamente esos trastornos cuando se presentan en esta etapa, teniendo en cuenta las posibles complicaciones y el peligro para la vida que pueden presentarse. El objetivo del presente artículo es proporcionar a los obstetras una revisión bibliográfica que les permita actualizarse acerca del tema y facilite su modo de actuación ante pacientes con coagulación intravascular diseminada. Se concluye que es preciso diagnosticar tempranamente el proceso en la fase bioquímica de bajo grado y aplicar en las féminas el tratamiento expedito de la enfermedad de base para eliminar las complicaciones o disminuir su ocurrencia.
Palabras clave: grávida, puérpera, hemostasia, coagulación sanguínea, coagulación intravascular diseminada.
ABSTRACT
In the period from pregnancy to puerperium, blood clotting suffers changes which are important to be known in order to interpret correctly those dysfunctions when they appear in this stage, keeping in mind the possible complications and the danger for life which can take place. The objective of the present work is to provide the obstetricians a literature review that allows them to be updated about the topic and facilitate their performance in case of patients with disseminated intravascular clotting. It is concluded that it is necessary to have an early diagnosis on the process during the low grade biochemical phase and to apply in these women the expedite treatment of the base disease to eliminate complications or to decrease their occurrence.
Key words: pregnant woman, puerpera, hemostasis, blood clotting, disseminated intravascular clotting.
INTRODUCCIÓN
Durante el embarazo y el puerperio se producen cambios en los mecanismos de la coagulación, entre los principales figuran: aumento de los factores de la coagulación, incremento de la actividad plaquetaria por agregación y depresión del sistema fibrinolítico
hacia el tercer trimestre, debido al aumento del inhibidor del activador del plasminógeno de tipos 1 y 2, así como disminución de su activador, lo cual facilita que las mujeres en esta etapa de la vida queden predispuestas con facilidad a la aparición de coagulación intravascular diseminada (CID) y de estados tromboembólicos.
Esta entidad es un mecanismo intermediario de múltiples afecciones obstétricas que se presentan con carácter agudo, subagudo o crónico y pueden conducir a la microtrombosis en la microcirculación y al daño de órganos. Según Metsuda, en etapa más tardía, la hemorragia complica el cuadro con la ocurrencia de choque que actúa como intermediario de las múltiples entidades de base. 1-3
En los últimos años han evolucionado los conocimientos relacionados con los mecanismos de la coagulación de la sangre, entre los cuales se encuentra la nueva comprensión de estos, teniendo en cuenta la alternativa que ha permitido esclarecer fenómenos que fueron desconocidos hasta la fecha; sin embargo, no es causal que las primeras descripciones e interpretaciones fisiopatológicas de la coagulación intravascular diseminada (CID) fueran descritas en pacientes obstétricas, que permitieron explicar estos nuevos mecanismos.1
Para lograr e interpretar adecuadamente los trastornos hemostáticos en el embarazo, es necesario conocer, no solo el funcionamiento de este sistema biológico y de sus desviaciones, sino también la necesidad de interactuar en su atención con hematólogos e intensivistas, que de conjunto con el obstetra, tendrán como objetivo brindar una atención calificada que permita realizar un diagnóstico precoz y reducir la mortalidad. 1, 3
Como bien se conoce, en la mayoría de estas pacientes el tratamiento de la enfermedad de base constituye una premisa indispensable en la resolutibidad del problema, de manera que el tratamiento con heparina solo estaría indicado en un número reducido de dichas pacientes, lo cual exige el dominio de sus indicaciones, dosis, vía de administración y momento adecuado para su empleo, teniendo en cuenta la relación riesgo- beneficio. De hecho, es obligación del obstetra estar a la altura de esta demanda, pues una heparina mal indicada puede agravar el cuadro y poner en peligro la vida de la paciente durante el período grávido-puerperal.1, 3, 4
Los elementos expuestos en esta revisión tienen como objetivo la comprensión y actualización del tema; si esto se cumple se habrá logrado el objetivo propuesto.
HEMOSTASIA NORMAL 1, 3, 5 - 8
Esta depende de la relación del vaso sanguíneo, de las plaquetas y de los factores de la coagulación. El organismo humano está preparado para la reacción en cada sitio y proporciona el control de las soluciones de continuidad presentadas constantemente en la vida cotidiana.
El vaso sanguíneo: Es considerado elemental en el proceso de la hemostasia, está dotado de contractilidad y resistencia para responder a la injuria. En el momento de la lesión se producirá una contractilidad inicial refleja que ralentiza el flujo de sangre para facilitar la participación plaquetaria en segundos, lo que se conoce como hemostasia primaria, seguida de la secundaria, que dura minutos y lleva a la formación del coágulo de fibrina; sin embargo, este fenómeno será localizado al sitio de la lesión, lo que evita la extensión y garantiza la recanalización vascular y reperfusión de órganos, todo lo cual es responsabilidad de los sistemas anticoagulante fisiológico y fibrinolítico, actividad que depende de la relación entre activadores e inhibidores.
Inicialmente, cuando ocurre la lesión de un vaso, se producirá un reflejo axónico que produce una vasoconstricción de carácter transitorio y se limitará a la zona de la herida. Esto permite ralentizar la circulación y facilita la interrelación entre el vaso, las plaquetas y los factores plasmáticos de la coagulación.
Son múltiples las funciones que se le conocen al vaso, particularmente al endotelio, que ya no solo es considerado un transportador de la sangre. Por ello, actualmente, en condiciones fisiológicas, se le concede una gran actividad de tromborresistencia, pero es capaz de
transformarse ante la presencia de trauma, hipoxia y otras injurias, en un endotelio que, basado en su gran plasticidad, podrá adoptar transformaciones defensivas limitadas al sito de la lesión, con la capacidad de reparar y restaurar la pared del vaso, así como también mejorar el flujo y reperfusión de tejidos y órganos afectados. Por tanto, esto implica que el endotelio vascular sea altamente activo y tenga sustancias coagulantes, las cuales actúan posteriormente en pro de la hemostasia y de la reparación vascular.
Con la participación del vaso se liberan luego sustancias como el tromboxano A2 y la serotonina, que producirán una vasoconstricción más sostenida, consecutiva a la primera señalada, de tipo neurógena.
Las plaquetas: Son elementos hemostáticos, no nucleados, de las que se ha podido identificar su estructura por medio de la microscopia electrónica. Se le define forma de disco, con un diámetro de 2 a 3 micras, rodeado de membrana y un citoplasma con mitocondrias, donde se lleva a cabo el metabolismo oxidativo. Estas se derivan de la fragmentación de los megacariocitos y, pese a ser fragmentos celulares, poseen una amplia maquinaria metabólica que les facilita cumplir con sus funciones hemostáticas. En su interior se presenta una zona granular central (granulómero) y otra periférica (hialórumo). La microscopia electrónica ha permitido identificar una membrana exterior que limita la zona sol-gel (citoplasma), un sistema canalicular y depósitos de glucógeno. En la zona gel aparecen 2 tipos de gránulos: los densos y los alfas.
En la circulación, las plaquetas se encuentran en estado no activado; la activación ocurre de la manera siguiente: por interacción de plaquetas con el endotelio, de plaquetas con plaquetas o del endotelio, células blancas y plaquetas. De forma básica, estas desempeñan una función importante, tanto en la hemostasia como en el proceso de reparación vascular.
Por otra parte, es conocido que la adherencia plaquetaria es la propiedad que tienen las plaquetas de adherirse al colágeno en el subendotelio, inmediatamente después de producirse el daño vascular. En esta adhesión participa un receptor glicoproteína Ia-IIa considerado un miembro de la familia de las integrinas. Más adelante, esta unión logra mayor estabilidad por medio de la participación del factor Von Willebrand, que une el receptor Ib- Ix a las fibras colágenas subendoteliales. Así, rápidamente, se produce la entrada de tromboplastina de la pared del vaso dañado hacia la luz vascular, lo cual genera la formación de una pequeña fracción de trombina, que participa junto a la epinefrina y la colágena como activadores de la fosfolipasa A2 y C, lo cual propicia la liberación de ácido araquidónico y la formación del tromboxano A2, considerado como un gran agregante plaquetario. Por otra parte, la hidrólisis de fosfolípidos de la membrana facilita el flujo de calcio dentro de las plaquetas, lo que contribuye al cambio de su forma discoide a la esférica, con seudópodos que permiten la interacción con otras plaquetas. El fenómeno de fosforilación de proteínas intracelulares determina la regulación de los diferentes gránulos: los densos segregan calcio, serotonina, adenosín difosfato (ADP), adenosín trifosfato (ATP), epinefrina y el factor plaquetario 4; los alfa, factor de crecimiento, von Willebrand, fibronectina y fibrinógeno plaquetario. Por último, la liberación del ADP produce cambios en las glicoproteínas IIb_IIIa que se unen al fibrinógeno y facilitan la unión de las plaquetas al tapón hemostático.
En el proceso de reparación vascular intervienen la actividad del factor de crecimiento, que estimula el crecimiento y migración de fibroblasto y las células musculares de la pared vascular.
Se conoce que la fosfatidilserina de la membrana interna plaquetaria se traslada a la porción externa cuando se activan las plaquetas, lo cual pone a los fosfolípidos en disposición de la activación de los factores de la coagulación, proceso conocido como flip-flop.
FACTORES PLASMÁTICOS DE LA COAGULACIÓN
Han sido determinados por el Comité Internacional de la Hemostasia y Trombosis. A estos se adjudicó un número romano como elemento de nomenclatura, aunque no tienen relación con su aparición en el proceso de la coagulación.
- Factor I. Fibrinógeno
Es una proteína plasmática con de peso molecular 340 000 daltones, que libera 2 monómeros con carga negativa, los cuales experimentan alineación terminoterminal y laterolateral y forman tiras de fibrina de unión laxa, fortalecidas posteriormente por la acción del factor XIII.
- Factor II. Protrombina
Es una alfa 2 glicoproteína, con peso de 68 000 daltones, fabricada en el hígado, de manera que necesita la vitamina K para su síntesis. Esta protrombina es transformada, por el factor conversor, en trombina enzima proteolítica de 35 000 daltones, necesaria para la transformación del fibrinógeno en fibrina.
- Factor III. Tromboplastina
Es una lipoproteína de alto peso molecular, con una porción proteica termolábil y una fracción lipídica termoestable, presente en la fracción microsomal de múltiples tejidos: cerebro, placenta y pulmón. De este factor solo se conocía su actividad en la vía extrínseca, pero hoy día se le concede gran importancia por su participación inicial en lo que se conoce como vía alternativa.
- Factor IV. Calcio
Participa prácticamente en todas las etapas de la coagulación; sin embargo, su déficit no es capaz de producir trastorno de la coagulación in vivo, pues sería incompatible con la vida la pequeña cantidad que se necesita en el proceso de la coagulación. Su participación es siempre en forma ionizada.
- Factor V. Proacelerina (Factor lábil)
Proteína termolábil que migra entre las globulinas alfa y beta. Su participación es necesaria en el factor conversor, tanto en la vía extrínseca como intrínseca.
- Factor VI. No existe
- Factor VII. Proconvertina (Factor estable)
Es el único identificado que participa en la vía extrínseca, se trata de una proteína producida en el hígado, es una vitamina K dependiente, con peso molecular de 15,000 a 25,000 daltones y migra entre las globulinas alfa y beta.
- Factor VIII. Globulina antihemofílica
Factor lábil del plasma, necesario para la conversión de la protrombina en el sistema intrínseco, con actividad deficiente en la hemofilia clásica. En las formas muy graves hay ausencia de factor VIII. Algunos señalan que los pacientes con enfermedad de von Willebrand poseen valores disminuidos de dicho factor.
- Factor IX. Componente tromboplastínico plasmático (PTC)
Es necesario en la actividad del factor conversor en el sistema intrínseco, es dependiente de la vitamina K y su déficit en sangre es considerado como productor de la hemofilia B.
- Factor X. Stuart- Prower
Alfa globulina con peso molecular de 87,000 daltones, se produce en el hígado en presencia de la vitamina K y participa como factor conversor de ambas vías.
- Factor XI. Antecedente tromboplastinico plasmático (PTA)
Emigra entre las globulinas beta y gamma, tiene un peso de 165 000 daltones y participa en la generación del factor conversor en el sistema intrínseco, pero no en el extrínseco.
- Factor XII. Hageman
Constituye el primero de los factores que se activan en la vía intrínseca, por cargas negativas de los grupos polares del acido glutámico y aspárgico de la colágena. In vitro es activado por el cristal; también acelera la actividad fibrinolítica y aumenta la dilatación y permeabilidad de los vasos.
- Factor XIII. Estabilizador de fibrina
Es una globulina plasmática activada generalmente por la trombina y tiene como función básica la estabilización del coágulo de fibrina, al producir fuertes puentes de unión gamma-glutamil-epsilon-lisina entre las unidades de fibrina, lo cual causa un coágulo fuerte.
Precalicreina (factor Fletcher) y quininógeno de alto peso molecular (factor Fitzgerald)
Intervienen en la fase inicial de contacto, pero está demostrado que la deficiencia de ambos, si se le conceden otras funciones biológicas, no produce diátesis hemorrágica.
Las explicaciones anteriores, para mejor su comprensión, fueron fundamentadas en el concepto clásico de las vías extrínseca e intrínseca; sin embargo, hay que dejar claro que esto ha variado en los últimos años, basado en un estudio profundo del sistema extrínseco y de forma particular en la interacción del complejo factor tisular-factor VII, con lo cual se ha logrado demostrar que este no solo activa el factor X, sino también actúa sobre el factor IX, componente de la vía extrínseca, lo que evidencia que ambas vías no son totalmente independientes. Estos elementos serán retomados al final y se iniciará la explicación de las vías conocidas inicialmente.
La formación de trombina constituye el momento cumbre en esta constelación de hechos; actúa como una enzima proteolítica sobre el fibrinógeno, de manera tal que lo escinde en los monómeros A y B, los cuales posteriormente, por la acción del factor XIII, transforma la malla tenue de dichos monómeros en otra firme y gruesa de fibrina. A su vez, la trombina se deriva de la protrombina elaborada en el hígado, como algunos factores en presencia de la vitamina K; esta transformación es un proceso complejo, que puede iniciarse en una de las vías mencionadas e identificarse en diferentes exámenes de laboratorio. Se sabe que ambas vías comparten los últimos pasos en el proceso de la coagulación, pero se diferencian en la forma de iniciarse.
Vía intrínseca
Se inicia con la activación del factor XII, que se favorece por la calicreína y con la participación de los quininógenos de alto peso molecular. In vivo, la exposición del colágeno subendotelial es básico en su estimulación, los otros elementos señalados coadyuvan a esta. El XIIa facilita la conversión del IX a IXa en presencia del calcio. Este último formará un complejo con el VIIIa y los fosfolípidos de la superficie de membrana en presencia de calcio, de modo que convertirá el X en su forma activa Xa, que unido al factor Va y el calcio forma el complejo conocido como protrombinasa, que convierte la protrombina en trombina, escinde al fibrinógeno en los fibrinopéptidos A y B y monómeros de fibrina, que posteriormente serán estabilizados por la actividad del factor XIII.
Vía extrínseca
Ha ganado importancia en los últimos años, en esta se inicia la formación de un complejo entre la tromboplastina tisular y el factor VIIa que actúa sobre el factor X y lo activa a Xa; a partir de este momento, se le concede el mismo mecanismo de la vía intrínseca. En sus inicios no se ha esclarecido la activación de pequeñas cantidades de VIIa, pero posteriormente el propio factor Xa facilita una mayor activación de VII a VIIa.
Vía alternativa
Sobre la base de múltiples investigaciones, se considera el complejo factor tisular-factor VII como el mecanismo más importante de la coagulación en vivo. Estos elementos se han basado en que la secuencia principal de activación de la vía extrínseca FVII-FX- FII, se verá reforzada por las reacciones ya conocidas FVII-FIX-FX, la cual se denomina vía alternativa (figura).
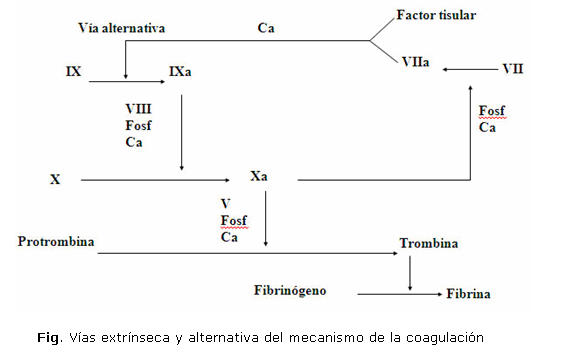
Fibrinólisis
Consiste en un sistema de respuesta que se inicia para contrarrestar la presencia de un coágulo de fibrina que necesita ser eliminado de la luz vascular para solucionar el flujo de sangre y garantizar la reperfusión de los tejidos comprometidos. De manera esencial, sin dicho sistema el organismo viviría en cuadros de trombosis permanente que serían incompatibles con la vida. Está regulado mediante un sistema de activadores e inhibidores.
Su precursor elemental es la presencia en plasma de una proenzima inactivada en condiciones fisiológicas (el plasminógeno), el cual se transforma en una enzima activa conocida como plasmina, capaz de digerir la fibrina, el fibrinógeno, hormona de crecimiento, corticotropina (ACTH) y otros factores de la coagulación, que en condiciones normales se encuentra en estado de inactividad.
El plasminógeno quedará activado por el predominio de sus activadores sobre los inhibidores. El activador tisular (tAP) es considerado el más importante, afirmación que se extiende al embarazo, además del activador del plasminógeno conocido como uroquinasa (u-PA). Ambos, al aumentar en plasma convierten el plasminógeno en plasmina, la cual actúa sobre la fibrina, es decir, la fragmenta y facilita su eliminación por el sistema reticuloendotelial.
También existe un sistema inhibidor de la fibrinólisis, dado por el inhibidor del activador del plasminógeno (IAP), así como los inhibidores de la plasmina, la alfa 2 antiplasmina y la alfa 2 macroglobulina.
El sistema fibrinolítico permite, además, que no se propague la formación del coágulo de fibrina más allá del sitio de la lesión.
Sistema anticoagulante fisiológico
Está constituido, en primer lugar, por la antitrombina III (ATIII), glicoproteína de peso molecular de 58,000 daltones, de síntesis hepática, considerada en el grupo de las serinproteasas, la cual se une a un cofactor heparínico endotelial y dicho complejo a la trombina y la neutraliza, así como a los factores XIIa, XIa, IXa, las calicreinas y la plasmina. La unión de la heparina con la antitrombina III conlleva a un aumento de la velocidad de interacción de la AT III con la trombina. Es importante conocer que las heparinas de bajo peso molecular tienen mayor acción, pues bloquean el factor Xa y con la heparina clásica predomina su efecto sobre la trombina. La concentración plasmática de la ATIII es de 0,8-1,2 unidades por mililitro. Según Fornier, los niveles disminuidos (por debajo de 70 %) se asocian a trastornos de la coagulación y se le concede a esta serinproteasa una vida media de 55 minutos.
La proteína C de producción hepática es otro anticoagulante fisiológico producida en el hígado, dependiente de la vitamina K. Esta se activa por la formación del complejo trombina-trombomodulina en el endotelio, de manera que la proteína C activada bloquea los factores Va y VIIIa y promueve fibrinólisis por bloquear el inhibidor del activador del plasminógeno de tipo I (PAI- 1), para lo cual necesita de la presencia de la proteína S que actúa como cofactor de la C. Desde el punto de vista clínico, el valor de esta última como cofactor, no quedó demostrado hasta que Griffin describió la primera familia con déficit congénito y la aparición de fenómenos tromboembólicos. Niveles decrecidos de proteína C y S se asocian a estados de hipercoagulabilidad.
MECANISMOS FISIOLÓGICOS DE LA COAGULACIÓN DURANTE EL EMBARAZO 1, 3, 4, 7, 9
Durante el embarazo se han observado cambios en la coagulación, entre ellos el aumento progresivo de los factores de la coagulación.
- Factores de la coagulación de mujeres embarazadas en tercer trimestre comparado a mujeres no embarazadas.
| Factores | Tercer trimestre | Controles |
| XIII | 76 | 100 |
| XII | 119 | 48 |
| XI | 86 | 48 |
| X | 148 | 100 |
| IX | 191 | 100 |
| VIII | 310 | 100 |
| VII | 190 | 100 |
| V | 169 | 100 |
| Protrombina | 202 | 100 |
| Fibrinógeno | 166 | 100 |
| Inhibidores | ||
| Antitrombina III | No cambia | 112 |
| Proteína C | 88 | 29 |
| Proteína S | Disminuye | 23 |
| Precalicreina | 200 | 64 |
| Kininógeno | 125 | 64 |
Durante la gestación, la sangre se describe como hipercoagulante, debido básicamente al aumento de los factores de la coagulación y, como también se plantea, de los complejos trombina-antitrombina y de los complejos solubles de fibrina, lo que muestra la activación de los mecanismos de la coagulación.
El incremento de la proteína C activada en el curso del embarazo, se interpreta como un mecanismo de compensación al aumento de factores como el Va y VIIIa. El inhibidor de la PCA, identificado como alfa-1 antitripsina, disminuye en el embarazo, lo que se explica por el aumento del consumo en respuesta a la mayor actividad de la PCA.
En estudios recientes se ha demostrado que durante el trabajo de parto, en la circulación uterina ocurre una CID de intensidad variable, debido a los niveles aumentados de tromboplastina tisular en relación con el lugar de inserción placentaria.
Cambios en las plaquetas durante el embarazo
Durante la gravidez, existe tendencia a la actividad trombótica; también se incrementa la agregación plaquetaria.
Los mecanismos en secuencia se interpretan de la manera siguiente:
- Aumento de la agregación plaquetaria
- Incremento de la prostaciclina en vasos maternos y fetales
- Disminución marcada de la sensibilidad de las plaquetas a la prostaciclina
- Disminución de la formación del monofosfato cíclico de adenosín (AMPc)
Se observa el aumento de la prostaciclina, sustancia antiagregante y vasodilatadora, pero se demuestra una disminución de la sensibilidad plaquetaria a esta en el tercer trimestre del embarazo (hasta en 30 %), lo cual explica la tendencia a la agregación plaquetaria.
Los niveles de AMPc disminuyen, estos tienen la propiedad de oponerse a la agregación plaquetaria y median los efectos de la prostaciclina. Esto demuestra falta de actividad del AMPc como mediador de enzimas como la adenilatociclasa y fosfodiesterasa.
Investigaciones recientes revelan que durante el embarazo normal el puntaje de plaquetas disminuye a partir de las 32 semanas hasta el parto, luego del cual se inicia su recuperación (en la semana 6 del posparto aproximadamente). Boehlen en un estudio comparativo con controles, en pacientes no gestantes, demostró que el percentil 2,5 de un adulto (en hombre o en mujer no embarazada), corresponde a 150 x 109/L; mientras que este mismo percentil, en mujeres embarazadas, corresponde a 116 x 109/L. De los anteriores planteamientos se deduce que durante la gestación existe una disminución fisiológica en el número de plaquetas, sin que represente alteración o aumento patológico del consumo de estas; sin embargo, estos cambios no se observan en la práctica médica con frecuencia.
Es conocida la relación existente entre conteo plaquetario y volumen medio plaquetario (VMP), el cual representa el tamaño de las plaquetas circulantes, descrito desde 1981, de tal forma que mientras menor es el número de plaquetas mayor será el VMP. De esto se desprende que durante la gestación existe una tendencia al aumento o no cambios del VMP. Durante el embarazo normal se reporta un mayor consumo plaquetario, aunque interpretado como compensado, y se muestran más cambios en la morfología plaquetaria.
De todas maneras, una interpretación adecuada es que durante el embarazo existe una mayor activación y consumo plaquetario, evidenciado por un menor número, aumento del volumen, agregados y metabolitos implicados en el incremento de la actividad plaquetaria, lo cual muestra un estado de hipercoagulabilidad conservada.
Cambios en el sistema fibrinolítico durante el embarazo
Este sistema representa el momento final del proceso de la hemostasia que resulta en un estado compensatorio con la restauración del vaso. El embarazo constituye un estado en el que se muestra disminuida la activación del sistema fibrinolítico, lo cual contribuye al estado de hipercoagulabilidad. Este cambio está relacionado con el aumento del inhibidor del activador de tipo 1 (PAI-1) y tipo 2 (PAI- 2). El primero se sintetiza en las células endoteliales, llega al término del embarazo con niveles de hasta 3 y media veces su valor basal; por su parte, el PAI-2, de origen placentario, alcanza hasta 30 veces su nivel basal al final de ese período. Por otro lado, se ha señalado disminución del activador tisular del plasminógeno, aunque en el embarazo se ha planteado aumento de los productos de degradación del fibrinógeno y del dímero D. El incremento de estos parámetros se detiene y en el tercer trimestre los complejos TAT ascienden, lo que demuestra en esta etapa una tendencia notable al estado de hipercoagulabilidad.
COAGULACIÓN INTRAVASCULAR EN OBSTETRICIA 1, 7- 23
La coagulación intravascular diseminada es un síndrome de origen multicausal que se caracteriza por la entrada en acción del sistema de la coagulación, dado básicamente por un aumento de la actividad de trombina que se genera intravascularmente y que se acompaña de un aumento de la activación de factores de la coagulación, así como de sus inhibidores naturales y una respuesta secundaria reaccional del sistema fibrinolítico.
Esta entidad clínica es considerada como una manifestación intermediaria de múltiples enfermedades en la que el daño tisular, como resultado del fenómeno trombótico, es la causa de la aparición consecutiva del daño múltiple de órganos. Puede aparecer de forma aguda y crónica. La primera ocurre cuando la actividad de los factores de la coagulación se presenta en escaso tiempo (minutos); en la segunda, la instalación es en días.
Es poco común referirse a formas localizadas y diseminadas, pero en realidad el proceso puede ser expresión intravascular en un órgano o manifestarse de forma diseminada, cuando afecta extensamente toda la microvasculatura. Una forma intravascular es planteada cuando existe un consumo de factores en la conformación, por ejemplo, de un gran hematoma.
Clasifican según Patogenia
De forma general, en 2 grandes grupos en los que se insertan también las causas obstétricas.
Tipo I. Las que se desencadenan por una activación en el sistema de la coagulación.
Tipo II. Las que se relacionan con un daño de la pared vascular.
En el primero existe básicamente una entrada en la circulación de sustancias procoagulantes, dada en múltiples enfermedades obstétricas por la entrada en la circulación de tromboplastina tisular. Es preciso recordar, que la placenta es un órgano rico en esta sustancia; otras fuentes pueden aparecer en órganos como cerebro y pulmón. La entrada de sustancias tóxicas al organismo como el veneno de serpiente puede también desencadenar el síndrome.
En el segundo puede aparecer lesión en la pared vascular, gran exposición de la colágena, así como daños inmunes con formación de antígeno anticuerpo en el endotelio e, incluso, generarse por la infusión de algunas aminas vasopresoras con efecto tóxico.
De manera directa, a continuación se señalan las causas fundamentales en obstetricia.
Tipo I. Infusión de sustancias con actividad tromboplástica
- Desprendimiento prematuro de la placenta
- Embolismo del líquido amniótico
- Síndrome de feto muerto
Tipo II. Causas inmunológicas. Vasculitis
- Preeclampsia_eclampsia
- Síndrome de HELLP
- Hígado agudo del embarazo
- Sepsis grave en obstetricia
Clasificación en obstetricia según la instalación clínica
- Aguda: Desprendimiento prematuro de la placenta, embolismo del líquido amniótico, preclampsia-eclampsia, síndrome de Hellp, hígado agudo del embarazo.
- Subaguda (por su forma de instalación): Sepsis grave en obstetricia (básicamente por microorganismos gramnegativos).
- Crónicas: Síndrome de feto muerto
Diagnóstico
Cuadro clínico: Inicialmente aparecen los síntomas de la enfermedad de base. Otras manifestaciones que pueden comprometer la vida de las pacientes son: la presencia de la microtrombosis en la microcirculación, con el consecutivo daño isquémico de órganos, así como la aparición del cuadro de hemorragia de apariencia "dramática" en muchos de ellas, por los niveles bajos de los factores de la coagulación, la disminución del número de plaquetas y la actividad secundaria del sistema fibrinolítico.
Las manifestaciones estarán determinadas por la etapa de la CID, pueden presentarse fenómenos de microtrombosis con daño de órganos, manifestado por insuficiencia renal con oligoanuria, dificultad respiratoria, estado confusional en el sistema nervioso central, necrosis hepatocelular en hígado y en corazón isquemia miocárdica.
El estado de choque, como señala Macay, representa un mecanismo intermediario en la presentación de la CID que, en obstetricia, generalmente acompaña a las hemorragias agudas procedentes del útero. También puede ocurrir sangrado múltiple (por heridas y mucosas, venipunturas, encías), así como grandes hematomas, entre otros.
Los fenómenos de trombosis de órganos pueden manifestarse con la frecuencia siguiente: daños renales (25 %), hepáticos (19 %) y pulmonares (16 %); choque (14 %), tromboembolismo pulmonar (7 %) y alteraciones del sistema nervioso central (2 %).
Etapas de la CID
Ha sido concebida en períodos de evolución subclínica hasta la aparición de las manifestaciones clínicas.
1. Período de activación
2. Período bioquímico
- Bajo grado (compensado)
- Alto grado (descompensado)
3. Período clínico
Período de activación
Es considerado compensado, pues surge como comienzo de la coagulación en determinados eventos y cirugías y son rápidamente controlados por mecanismos compensadores fisiológicos de la propia coagulación(anticoagulantes fisiológicos), que generalmente no resultan en CID.
El hallazgo de un tiempo de cefalina kaolín acortado, que se puede encontrar en gran parte después de la cirugía mayor, sobre todo cardiovascular, indica este estado de activación. Se observarán factores activados con niveles de concentración plasmática normales, ligero aumento del fibrinógeno y del número de plaquetas, comparado con valores prequirúrgicos; estos estados no necesitan tratamiento.
Período bioquímico
- Período de bajo grado: Determina una forma evolutiva de la enfermedad, todavía sin compromiso de órganos, pues no hay bloqueo de la microcirculación y es el momento ideal para iniciar la terapéutica. Algunos autores lo denominan de hipercoagulabilidad o preclínico. En esta etapa el moderado consumo de los factores de la coagulación estará compensado por una célula hepática aún funcionante y el nivel de los factores no estará alterado sustancialmente.
- Período de alto grado: En este se establece el compromiso de órganos que puede ser moderado o severo y significa falla funcional, puede abarcar múltiples órganos, tales como: riñón, pulmón, hígado, intestino, corazón y cerebro.
En este período bioquímico, de alto grado o descompensado, el estado clínico del paciente comienza con un deterioro paulatino y es característico de los cuadros agudos, por lo cual es necesario realizar estudios seriados de la coagulación a corto plazo. Se añade que al estar comprometido el hígado, se perderán los mecanismos compensatorios y disminuirán los factores de la coagulación.
Resulta fácil suponer que en una enfermedad aguda que se presenta en pocas horas, muchas veces es difícil definir límites entre los diferentes períodos, por lo cual ante una causa potencial de CID, el estudio periódico de la coagulación permitirá orientar el diagnóstico y el tratamiento.
Período clínico
Generalmente se presentan cuadros de hemorragia (hematemesis, metrorragias, hemoptisis, entre otros). Por otro lado, también puede existir daño de órganos, dado por alteración funcional en riñón (anuria), pulmones (dificultad respiratoria), cuadros confusionales cerebrales, hepáticos (necrosis hepatocelular) e isquemia miocárdica, por citar algunos.
En el curso de la CID hay que tener en cuenta el fenómeno reaccional que se produce dentro del endotelio, como respuesta a la microtrombosis en la microcirculación, dado por la participación de la fibrinólisis secundaria cuando predominan los activadores de este sistema, activador tisular y uroquinasa (tPA y UPA) que sobrepasan los inhibidores (PAI-I), lo que puede facilitar la reperfusión de los tejidos y órganos por la recanalización vascular. Metsuda señala que en las manifestaciones clínicas de la CID el grado de fibrinólisis que acompaña a la enfermedad desempeña una función importante. El incremento de la actividad fibrinolítca no causa daño orgánico, este se produce cuando esa actividad es inadecuada. El fenómeno fibrinolítico es reaccional a los estados de trombosis y en muy pocas ocasiones se excede como para necesitar el uso de antifibrinolíticos.
Métodos sensitivos de diagnóstico. Marcadores de trombosis
Yamamoto y Saito describieron métodos de diagnósticos considerados como marcadores moleculares, entre los cuales figuran:
1. Fragmento 1+2, este es producto de la transformación de la protrombina en trombina en la porción aminoterminal.
2. Proteasas y complejos que se forman con los inhibidores. Complejo trombina-antitrombina III (TAT), otros IXa-ATIII, Xa-ATIII, plasmina-antiplasmina (PAP).
3. Fibrinopéptido A y B
4. Monómeros de fibrina y degradación fibrinógeno-fibrina (PDF- pdf)
5. Fibrina polimerizada. Dímero D.
El fragmento 1+2, el fibrinopéptido A y B, los complejos TAT y monómeros de fibrina son marcadores de la presencia de trombosis en la circulación. Definen el diagnóstico precoz.
El complejo plasmina antiplasmina (PAP), los PDF-pdf y el dímero D, reflejan actividad plasmática, diagnóstico tardío de enfermedad avanzada.
La posibilidad del diagnóstico depende del nivel de activación de la coagulación y de la posible sensibilidad de los diferentes medios de diagnóstico. Por eso una interpretación coherente está dirigida a que un solo medio de diagnóstico no puede descartar la presencia de CID, como tampoco debe certificarla.
Formas clínicas de CID en obstetricia
Desprendimiento prematuro de la placenta normoinserta: Es la forma más común de CID en obstetricia y presenta frecuencia de 0,5 a 1,3 % de las embarazadas. La patogenia de esta enfermedad no está claramente definida, aunque se señala la participación de hipoxia debido a microinfartos placentarios. Esta afección puede ocurrir en pacientes aparentemente normales o en gestantes con preeclampsia. La severidad del daño hemostático se relaciona, por lo general, con el grado de desprendimiento placentario. El fallo hemostático puede estar relacionado con la presencia de sangrado proveniente del útero, así como también de la pérdida de sangre por otras mucosas, venipunturas, entre otras.
Buettner señala, en pacientes con hematoma retroplacentario, la aparición de desprendimiento de retina por oclusión de vasos coroides, como expresión de microtrombosis que acompaña a la CID.
El diagnóstico de desprendimiento prematuro de la placenta es eminentemente clínico y se corrobora por ecografía si fuera necesario; asimismo, la identificación de la CID se realiza con los medios de diagnósticos correspondientes y se condicionará la solución de esta entidad a la terapia primaria que es la evacuación inmediata del útero, pues cuando se alcanza la contractilidad miometrial eficiente es capaz, por sí sola, de corregir el trastorno. La terapia de reposición de factores con plasma fresco y factores, es de gran importancia para revertir el cuadro.
En esta entidad no existe indicación para la heparina. Algunos autores han defendido el tratamiento antifibrinolítico, aunque es controvertido.
Hallazgos de CID en el desprendimiento prematuro de la placenta
| Aumentan | Disminuyen |
| - Fibrinopéptido A | - Plaquetas (trombocitopenia) |
| - Complejo TAT | - Factores coagulación |
| - ATIII | |
| - Dímero D |
|
Muerte fetal intraútero
Los cambios hemostáticos que se producen tienen que ver con la duración de la retención. Si esta es de 5 semanas, ocurre una profunda hipofibrinogenemia en 40 % de las pacientes. Para algunos, el trastorno es básicamente una hipofibrinogenemia selectiva, que en ocasiones se acompaña de trombocitopenia.
Aunque su causa es algo indeterminada, lo más aceptado es que existe una baja, pero continua producción de sustancias tromboplásticas procedentes del feto y la placenta, que pasa a la circulación y ocasiona un tipo particular de CID, considerada como crónica. Generalmente se presenta hipofibrinogenemia y, en ocasiones, la presencia de productos de degradación de la fibrina. Después de confirmado el diagnóstico, la conducta básica es la evacuación rápida del útero. En ocasiones, la caída del fibrinógeno deberá acompañarse de su reposición con crioprecipitado para mejorar los valores y proceder a la evacuación uterina.
Este tipo de coagulopatía puede responder al uso de heparina, lo cual prevendrá la actividad de trombina por la entrada de la tromboplastina en la circulación, aunque su uso es cuestionado por quienes plantean que el cuadro revierte solo con el tratamiento de la enfermedad de base. Cuando se utiliza la heparina, debido a su corto plazo de vida, puede ser descontinuada en el momento del parto y no requerir reversión de rutina.
Hallazgos de CID en el síndrome de feto muerto
| Aumentan | Disminuyen |
| - Monómeros de fibrina | - Fibrinógeno |
| - Fibrinopéptido A | - Plaquetas (trombocitopenia) |
| - TAT | - Factores coagulación |
Infección obstétrica grave (estado de choque séptico)
Puede estar asociada con aborto séptico y con infección intrauterina anteparto o posparto. La infección por microorganismos gramnegativos es el agente más frecuente, determinado por la acción de las endotoxinas, la estimulación del sistema de la coagulación, el complemento y las quininas; todos estos cambios facilitan la aparición de la CID.
Generalmente, los trastornos de la coagulación que se presentan en la CID, se acompañan de depósitos de fibrina en la circulación y se relacionan con la insuficiencia multiorgánica.
Desde el punto de vista fisiopatológico es conocida la función de las endotoxinas, básicamente gramnegativos de alto peso molecular (1 millón de daltones) que desencadenan la CID, lo cual ha podido demostrarse con claridad en modelos animales. El fenómeno de Swartzman, en conejos, deja claro el daño que se produce después de la inoculación de la toxina en el riñón, lo cual ocasiona necrosis cortical bilateral y trombosis glomerular marcada, de manera que es considerado el daño más característico en estos casos.
Estudios recientes corroboran que las endotoxinas, fundamentalmente polisacáridos, son los desencadenantes del estado de choque endotóxico, por medio de la estimulación de macrófagos, monocitos o ambos, con la consecutiva liberación de citoquinas, entre las cuales figuran: el factor de necrosis tumoral (FNT), el interluquin-1 (IL-1), interluquin-6 (IL-6), el interluquin-8 (IL-8) y el interferón alfa (á-IFN). De todas estas citoquinas, el rol fundamental en el desencadenamiento de la septicemia se le concede al FNT, pues se ha demostrado que este actúa sobre polimorfonucleares y sobre el endotelio vascular, lo cual permite activar los receptores de adhesión CDW 18, participantes en la unión de los polimorfonucleares entre sí y activa el ELAM-1, que favorece la quimiotaxia y facilita la adhesión de estas células al endotelio.
El FNT participa en el desencadenamiento de la CID de la manera siguiente:
1. Se inicia la activación de la CID en los procesos sépticos por la actividad del FNT que se encuentra en la adventicia de la pared vascular.
2. Estimula la síntesis del FT, lo cual se favorece por el aumento de la permeabilidad del endotelio, de modo que facilita la actividad del complejo iniciador de la coagulación FT-FVII.
3. Actúa sobre el sistema fibrinolítico, expresado en una disminución de su actividad. Se debe al aumento de la síntesis del inhibidor del activador del plasminógeno (PAI-I) y por la disminución del activador del plasminógeno t-PA, cuando por otro lado se mantiene aún la activación de la coagulación, que determina un estado de hipercoagulabilidad.
4. Disminuye la actividad de la trombomodulina en el endotelio, por lo que no se formará el complejo trombina-trombomodulina que estimula proteína C y bloquea factores Va y VIIIa.
5. Favorece la síntesis del factor estimulante de plaquetas que produce un aumento de la agregación plaquetaria.
Hallazgos de CID en las pacientes obstétricas con sepsis grave
| Aumentan | Disminuyen |
| - Monómeros de fibrina | - Factores vitamina K |
| - TAT | - Fibrinógeno |
| - Frag 1+2 | - Otros factores |
| - Fibrinopéptido A | - ATIII |
| - Monómeros de fibrina | - Trombocitopenia |
| - Dímero D | |
Resulta básico en el tratamiento el uso de antimicrobianos, la evacuación uterina y las medidas generales de sostén, dada principalmente por la corrección volumétrica. El uso de heparina para corregir el trastorno es discutido en la práctica general, pero en obstetricia la tendencia es a su empleo con lo cual en nuestra experiencia se presentan resultados favorables.
Embolia del líquido amniótico
El síndrome es un accidente dramático en el que se presenta una primera etapa de insuficiencia ventricular izquierda, seguida de una segunda etapa de CID y la tercera de daño de órganos, esta última relacionada en muchos casos con eventos de microtrombosis.
En la embolia del líquido amniótico aparecen:
1. Exceso de actividad fibrinolítica que refleja un aumento del plasminógeno.
2. Elevación de plasmina o alta concentración de PDF
3. Disminución de factores plasmáticos de la coagulación
Es importante destacar que las pruebas de laboratorio, compatibles con coagulopatías, pueden encontrarse alteradas de 30 a 60 minutos antes de las manifestaciones clínicas de CID.
Los mecanismos fisiopatológicos básicos en la CID que acompañan esta entidad son:
1. La entrada de gran cantidad de sustancia tromboplástica en la circulación (Hesneider_Hazteld)
2. Lesión endotelial con excesiva exposición del colágeno por la toxicidad del líquido amniótico (Heisner)
3. El mucus del líquido amniótico activa el factor X y desencadena el mecanismo de la coagulación (teoría de Phillip y Davidson).
Aunque el líquido amniótico carece de plasmina y de su activador, se conoce su riqueza como proactivador de plasmina, por lo que puede resultar en una coagulopatía dominada por fibrina-fibrinogenolisis. Se ha señalado que la contractilidad uterina se verá comprometida por los altos niveles depositados de PDF en los hiatos de la fibra muscular, aparece entonces la atonía uterina y la franca hemorragia.
Se ha sugerido que es la fibrinogenolisis primaria, y no la secundaria por CID, la causa de hipofibrinogenemia asociada a embolia del líquido amniótico.
Hallazgos de CID en la embolia del líquido amniótico
| Aumentan | Disminuyen |
| Factor Xa | PAt |
| PAI-1 | Factores de la coagulación |
| PAI-2 |
|
Preeclampsia-síndrome de HELLP
Como bien se conoce, la preeclampsia afecta aproximadamente el 10 % de los embarazos y constituye una causa importante de morbilidad y mortalidad materna y perinatal, predominantemente en países en desarrollo. Esta afección, denominada enfermedad de las teorías, durante años ha sido motivo de múltiples controversias científicas.
La afirmación aceptada es en cuanto a la presencia de un proceso de placentación anormal, pues de forma fisiológica el trofoblasto se divide en 2 porciones: la interna (citotrofoblasto) y la externa (sincitotrofoblasto), esta deberá erosionar la decidua uterina y penetrar para implantarse en el útero y tomar la sangre de los sinusoides de las arterias espirales uterinas.
Este fenómeno tiene una primera fase alrededor de la segunda semana, con penetración de la decidua, y una segunda fase que ocurre entre las 15 y 20 semanas, con penetración del trofoblasto en arteras espirales. Todo esto garantiza el flujo adecuado entre ambos compartimientos y el aseguramiento de un sistema protector de bajas presiones.
Asimismo, en la preeclampsia ocurre un defecto de penetración del trofoblasto en el curso de la placentación, lo cual genera invasión inadecuada y oclusión consecuente de capilares, de modo que conduce a la isquemia placentaria, determina hipoxia y liberación de citoquinas IL-1, IL-6, que pasan a la circulación y generan daño de las células endoteliales en la placenta y a nivel sistémico. Esta disfunción endotelial genera la disminución del óxido nítrico y de prostaglandinas vasodilatadoras, así como el aumento de endotelinas, serotonina y procoagulantes. Esta disfunción endotelial tiene como consecuencia fundamental, vasoconstricción generalizada, hipoperfusión de órganos y coagulación intravascular diseminada. Por tanto, puede aseverarse que en la preeclampsia existe una disfunción endotelial sistémica.
Las investigaciones realizadas en el colegio panamericano para el estudio del endotelio, han aportado grandes avances en este campo, pues no solo es el conductor de la sangre como se pensaba. Se plantea que es una interfase unicelular dinámicamente estable, mutable y bioquímicamente activa entre el torrente circulatorio y la intimidad de los tejidos. El endotelio es, además, un órgano multifuncional y de gran plasticidad, que reposa sobre una membrana basal sintetizada por ella misma en contacto con la íntima capa de linaje hemopoyético, cuya población mesenquimal indiferenciada se encuentra inmersa en una matriz extracelular. Esta población mesenquimal es capaz de producir macrófagos, linfocitos, dendrocitos, mastocitos, fibroblastos y células de la musculatura lisa. La matriz extracelular subendotelial es una superficie trombogénica que favorece la adhesión de plaquetas y la activación de la coagulación.
El endotelio responde a muchos genes activadores o desactivadores, que intervienen en el proceso salud _ enfermedad y propician la aparición de enfermedades, tales como: hipertensión arterial, ateroesclerosis y síndrome de HELLP; además de ello:
1. Se conoce como el órgano regulador de la presión sanguínea.
2. Forma parte del team de la hemostasia.
3. Actúa como compañero sparring de varios tipos de células sanguíneas.
4. Es pareja de baile de las células de la musculatura lisa vascular.
5. Origina hormonas, factores de crecimiento, receptores endoteliales, radicales libres de oxígeno, sustancias vasoactivas y factores hemostáticos.
Teniendo en cuenta que se parte de un daño endotelial placentario y sistémico, básicamente es posible percatarse del significado de la placenta como favorecedora de la ocurrencia de esta entidad, si se le reconoce como un órgano esencialmente constituido de endotelio; de hecho, si el endotelio es el órgano regulador de la presión sanguínea, se producirá un desbalance en la producción de sustancias vasoactivas con predominio de los FEC (angiotensina, tromboxano, prostaglandina H y endotelina), sobre los FER (prostaciclina, factor repolarizante del endotelio, oxido nítrico).
Lo anterior lleva a hipoxia, a la liberación de radicales libres y al daño del endotelio, se invoca un aumento de la actividad de moléculas de cohesión intracelular (ICAM) y de las de adhesión intravascular (VCA), en valores de 350 y 1 300 (µg/mL), respectivamente, así como una disminución de vitaminas antioxidantes que perpetúan el daño, donde se podrá activar la coagulación, clasificado como CID de tipo II, teniendo en cuenta su origen.
Considérese básico el mecanismo activador del factor XII, partiendo del daño subendotelial como esencial de esta enfermedad, además, de la actividad plaquetaria; sin embargo, todavía hay autores que defienden su asociación con la vía extrínseca y argumentan la posibilidad de activación tromboplástica por la placenta, mecanismo menos aprobado en la actualidad.
Un hecho de importancia lo constituye la transformación plaquetaria en el curso de la preeclampsia, dado por puntaje de plaqueta bajo, incremento de volumen y aumento de microagregados plaquetarios, lo cual evidencia en el criterio de algunos autores que estos cambios pueden preceder al inicio de la enfermedad; también se ha demostrado un aumento de la actividad de la tromboglobulina (factor plaquetario 4), considerado como expresión de actividad plaquetaria al relacionarse con los niveles de proteínas de 24 horas. Se plantea que esta entidad cursa con un aumento del consumo plaquetario, a lo que contribuye el desbalance entre el tromboxano, la prostaciclina y el óxido nítrico, con predominio del poder agregante del primero; para algunos, el daño plaquetario es desencadenante y no resultado. Además, la alteración de la circulación placentaria es motivo de estimulación plaquetaria, a través del aumento de estrés de fricción, producto de invasión trofoblástica inadecuada con flujo arterial anormal (notches).
La disminución de la ATIII es considerada una de las alteraciones básicas en esta enfermedad. El mecanismo del déficit no está esclarecido, pudiera ser aumento de consumo o disminución de producción hepática, por el daño de este órgano que acompaña a esta entidad clínica.
El comportamiento del sistema fibrinolítico ha sido muy controvertido en la preeclampsia. Unos refieren incremento de la fibrinólisis consecutivo a CID, dado por aumento de los PDF y disminución del plasminógeno y alfa 2 antiplasmina; otros señalan depresión de la actividad fibrinolítica debido a que aumentan los inhibidores del activador del plasminógeno.
Es válido destacar que el aumento de la concentración de calcio intracelular en esta entidad, es básico para incrementar la activación plaquetaria, relacionada, además, con niveles disminuidos del AMPc.
La peroxidación lipídica que acompaña a la preeclampsia favorece la permeabilidad capilar con aparición de edema y proteinuria, pero también desencadena trombosis por aumento de la actividad de trombina y liberación del inhibidor del activador del plasminógeno. La lesión en capilares de los glóbulos rojos, con la aparición de esquistocitos, observados en lámina periférica, dan crédito al daño endotelial como el disparador de los eventos relacionados con CID.
Hallazgos de CID en preeclampsia y síndrome de HELLP
| Aumentan | Disminuyen |
| - Fibrinopéptido A | - Fibrinógeno |
| - Monómeros de fibrina | - ATIII |
| - TAT | - Proteína C |
| - Tromboxano | - Precalicreína |
| - Plasminógeno | |
| - Calcio intracelular | - α antiplasmina |
| - β tromboglobulina | - Prostaciclina |
| - PAI | - Trombocitopenia |
|
| - AMPc |
PRUEBAS CLÁSICAS DE COAGULACIÓN EN LA CID 1, 2, 6, 8, 16
1. Tiempo de protrombina (tiempo de Quick): Su prolongación indica disminución de los factores I, II, V, VII y X.
2. Tiempo de tromboplastina parcial activado (TPTA-kaolín): Cuando se prolonga indica descenso de factores, básicamente V y VIII, los restantes se consumen en menor medida, no incluye factor VII.
3. Tiempo de trombina: Se altera cuando disminuyen los niveles plasmáticos de fibrinógeno, en presencia de productos de degradación del fibrinógeno o de heparina circulante.
4. Recuento plaquetario: Disminuye por secuestro en el coágulo, posee una alta sensibilidad para el diagnóstico de la CID, cercano a 90 %. En etapa avanzada de la enfermedad es útil para controlar la evolución.
5. Dosificación de fibrinógeno: Su disminución es de poca sensibilidad para el diagnóstico de CID; sin embargo, posee alta especificidad, cerca de 80 %. Su descenso se considera tardío y tiene valor para seguir la respuesta al tratamiento.
6. Productos de degradación de fibrinógeno (PDF), de la fibrina (pdf) y de la fibrina entrecruzada o polimerizada (dímero D): Estos detectan aumento de la actividad de plasmina, se incrementan con el aumento de la actividad fibrinolítica, tanto sobre el fibrinógeno como la fibrina.
La determinación de los PDF ha sido desplazada por el dímero D, un método más sensible que se puede determinar por pruebas inmunológicas cuantitativas (ELISA) o cualitativas (LATEX). Es considerado uno de los marcadores más precoces de CID y con alta sensibilidad (92-100 %). Valor normal: 0,5-0,8 microgramos por mililitro.
7. Dosificación de factores de la coagulación: Particularmente se dosifican los factores V y VIII, estos permiten detectar cambios de forma precoz por resultar muy sensibles para detectar la presencia de consumo; también resulta importante el ascenso de sus valores como repuesta al tratamiento precoz.
8. Antitrombina III (ATIII). Valores normales: 0,8-1,2 unidades por mililitro, niveles decrecidos, de 70 %, son compatibles con CID.
9. En la CID de tipo II se pueden observar esquistocitos en lámina periférica, aumento de la bilirrubina indirecta y de la deshidrogenada láctica (DHL), con disminución de la haptoglobina.
Recomendaciones
- Los niveles de fibrinógeno y factor VIII pueden estar aumentados en las etapas iniciales de la CID, por ser reactantes de fase aguda, aunque luego son consumidos.
- El control evolutivo es recomendable mediante evaluación de: TPT, determinación de niveles de fibrinógeno, número plaquetas y dosificación de factores V y VIII.
- La determinación del número de plaquetas, dímero D, monómeros de fibrina pdf y concentración de ATIII, se consideran las pruebas más confiables para el diagnóstico de CID.
Diagnóstico diferencial
1. Coagulopatía dilusional postransfusional: Esta adquiere consideración cuando en menos de 3 horas se ha administrado 50 % o más de la volemia estimada, lo cual se acompaña de plaquetopenia y descenso de los factores de la coagulación.
2. Coagulopatías congénitas: Se presentan con el déficit específico a un determinado factor, en muchos casos puede ser conocido el antecedente.
3. Fibrinólisis primaria: Su presentación es poco común, pues casi siempre que se presenta este cuadro existe una fibrinólisis secundaria. Para su diagnóstico puede ser elemental la presencia del número de plaqueta normal, por la ausencia de fase trombótica. El dímero D no se elevará.
4. Insuficiencia hepática con déficit de factores de la coagulación
5. Púrpura trombocitopénica trombótica y síndrome hemolítico urémico: Estas entidades se caracterizan por la presencia de plaquetopenias y anemia hemolítica. La primera se acompaña de trastornos neurológicos; la segunda, en muchos casos, se inicia en el puerperio. Los factores de la coagulación pueden modificarse poco o no mostrar cambios.
TRATAMIENTO 1, 2, 13, 16-36
1. Tratar la enfermedad de base
2. Reponer el déficit de factores
3. Anticoagulantes
4. Antifibrinolíticos
Teniendo en cuenta que en los eventos obstétricos el tratamiento de la causa desencadenante puede solucionar la CID, esto debe ser una prioridad. El problema puede ser resuelto mediante la cesárea en un desprendimiento prematuro de la placenta, o la evacuación uterina en un feto muerto con trastorno de la coagulación o el uso enérgico de antimicrobianos y la histerectomía obstétrica en las pacientes que requieren la extirpación del útero, para eliminar toda fuente de actividad procoagulante. En presencia de daño inmunológico y vasculitis, la protección de los endotelios es fundamental; el uso de los corticoides y la terapia antioxidante pueden desempeñar una función decisiva.
En los casos en que se conserva una función hepática adecuada, la regeneración de factores vitamina K dependiente, puede ser por sí sola determinante en el control del cuadro después de la corrección de la causa básica.
Reposición de volemia y factores
Resulta elemental la vigilancia de los signos vitales para la detección temprana de trastornos hemodinámicos. Se recomienda, unánimemente, la no reposición de factores cuando estos se encuentran bajos, si no hay hemorragia ni se van a realizar procederes invasivos.
En casos de hemorragia y déficit de factores se seguirán las pautas siguientes:
1. Mantener, en lo posible, la normovolemia o trabajar sobre el concepto de hipotensión permisiva
2. Lograr valores de hematocrito entre 25 y 30 vol %
3. Plaquetas por encima de 20 000 por mm3
4. Valores de fibrinógeno por encima de 100 mg/dL
En presencia de cirugía de urgencia aún sin sangrado, la reposición de factores y plaquetas deberá alcanzar los valores siguientes:
1. Tiempo de protrombina o de Quick mayor de 50 %
2. TPT dentro de límites normales
3. Factores con niveles de su actividad mayores de 50 %, de 0,5 Ud/mL
4. Número de plaquetas por encima de 50, 000 por mm3
Plasma fresco
Aporta la mayoría de los factores de la coagulación. Se debe administrar 1 unidad por cada 15 kg de peso. Cuando existe disponibilidad y hay riesgo de descompensación cardíaca con la administración de volúmenes de plasma se pueden utilizar los concentrados de factores vitamina K dependiente.
Los paquetes de plasma son de 200 o 250 mL, se dice que la administración de 6 a 8 paquetes repone 50 % del volumen plasmático y aumenta en 30 - 40 % los factores de la coagulación.
Deberá administrarse 1 paquete de plasma por cada 4 o 5 de glóbulos o sangre completa almacenada.
Crioprecipitado
Producto del descongelamiento rápido del plasma a 4°C, centrifugado y recongelado.
En la paciente obstétrica debe administrarse 1 unidad por cada 6 kg de peso. Este aporta 250 mg de fibrinógeno, 80-100 unidades de factor VIII, así como factor von Willebrand. Este se administrará con el objetivo de mantener valores por encima de 100 mg/dL (cada unidad eleva el fibrinógeno en 6-8 mg); unas 10 bolsas aportan aproximadamente 2 gramos de fibrinógeno que deben corregir una hipofibrinogenemia importante.
Concentrado de plaquetas
Deberá administrarse 1 unidad por cada 10 kg de peso, se indicará ante una plaquetopenia menor de 20 000 por mm3, cuando los valores son menores de 50 000 por mm3 y si se va realizar cirugía o procederes invasivos. Cada unidad eleva el conteo plaquetario en 5 000 a 10 000 por mm3. Por cada 6 a 10 paquetes que se administren se alcanzan valores de 50 por 109/ L.
Concentrados de ATIII
Actualmente es considerado uno de los tratamientos exitosos en la etapa inicial de la CID, así como el principal inhibidor natural de la trombina. La dosis terapéutica es de 90 a 120 Ud/kg y se continúa con infusión de 20 a 30 Ud/kg/día durante 4 días. Por cada Ud/kg de peso administrada el nivel plasmático aumenta en 1%.
Preparados vitamina K dependiente
Estos preparados aportan factor II, IX y X en 600 unidades y el factor VII, 500 unidades. Los requerimientos son de 1Ud/ kg de peso.
ANTICOAGULANTE. HEPARINA 1,2, 17, 24- 28, 33, 35-39
La terapia anticoagulante adquiere en obstetricia sus particularidades en el curso de la CID.
Deberá administrarse básicamente en la etapa de hipercoagulabilidad, cuando existe posibilidad de un diagnóstico temprano y en aquellas situaciones donde hay fenómenos trombóticos con sospecha de compromiso de órganos que acompaña a la CID. Este concepto no debe confundirse con el de enfermedad tromboembólica que puede aparecer en el embarazo, donde la terapia antitrombótica es la piedra angular del tratamiento.
Como principio general, no se aprueba el uso de heparina en presencia de sangrado, aunque algunos la utilizan. Las razones a esta objeción están dadas por varias razones: en primer lugar, en las hemorragias obstétricas se pierde rápidamente la ATIII, que es una glicoproteína a la que se une la heparina en una porción pentasacárida de su molécula, para bloquear trombina y otros factores de la coagulación. Por tanto, será sensato esperar la reposición mediante la administración de plasma o de ATIII en los lugares de disponibilidad para luego poder indicar la heparina; en segundo lugar, es preciso tener en cuenta que en un número importante de pacientes la hemorragia obstétrica se acompaña de estado de choque y que cuando este aparece, la hipoxia y la acidosis se instalan rápidamente. Es preciso recordar que la heparina no actúa cuando disminuye el PH, pues queda inactivada.
Aunque fue descrita en 1916 por Mc Lean, no fue hasta 1937 cuando se utilizó en un paciente con embolismo pulmonar. Está constituida por cadenas de polisacáridos compuestos por glucosamida, ácido idurónico y glucurónico en secuencias repetidas. Se obtiene comercialmente del intestino porcino o del pulmón bovino y son preparaciones heterogéneas. Se considera el anticoagulante ideal durante el embarazo, pues no atraviesa la placenta.
La acción antitrombótica de la heparina se debe fundamentalmente a su capacidad de potenciar la de la antitrombina (ATIII), a la que se une mediante una fracción pentasacárida específica, que se encuentra solo en un tercio de la cadena de HNF. La ATIII es considerada una inhibidora serinproteasa que bloquea la trombina y factor Xa y, en menor cuantía, otras enzimas. La presencia de heparina produce un cambio estructural en la ATIII, lo cual ocasiona una mayor actividad neutralizante, pues potencia 1 000 veces más su acción ante una concentración de heparina de 1 Ud/mL.
Para inactivar la trombina se requieren 13 unidades de sacáridos y para la inhibición del factor Xa solo 5. Todas las heparinas no fracciondas (HNF) contienen como mínimo 18 sacáridos y un peso promedio de 15 000 daltones.
La heparina ejerce otras acciones antitrombóticas de menor cuantía, tales como: potenciar su propio cofactor II, inducir actividad fibrinolítica por liberación del activador tisular del plasminógeno y cierta capacidad para inhibir la función plaquetaria. Clasifica como el antitrombótico ideal cuando se requiere lograr una rápida anticoagulación, pero toda vez que se conocen algunas de las limitantes de la HNF es obligatorio considerar su uso en el medio hospitalario.
De hecho, la cinética de eliminación es compleja mediante un mecanismo raudo y saturable de captación por células endoteliales, aunque existe otro más lento, pero de primer orden (de tipo renal), lo cual hace que su vida media varíe en dependencia de la dosis; por otro lado, tiende a unirse a proteínas plasmáticas, de manera tal que conduce a una gran diversidad de respuestas de un individuo a otro y justifican la necesidad de un control estricto.
Las heparinas se comercializan en forma de sales sódicas y cálcica, las cuales se administran por vía endovenosa y subcutánea, respectivamente; aunque esta última tienen una acción más lenta. Generalmente se controla la terapéutica con HNF mediante el TPTA, que se tratará de mantener alrededor de 1,5 - 2,5. Se presenta en bulbos de 5 mL (1 mL=50 mg=5 000 Ud).
Se plantea que la longitud de las cadenas de heparina no influye en la potenciación del efecto de la antitrombina sobre el factor Xa; sin embargo, en la acción inhibidora de la trombina es importante la presencia de cadenas largas, que pueden unirse, a la vez, a la ATIII y a la trombina, hasta formar un complejo terciario.
En la sepsis grave en obstetricia, el criterio de uso de heparina para detener la CID es discutido, aunque la mayoría están a favor de su utilización; los que no defienden su empleo
señalan que en la sepsis y el estado de choque el tratamiento adecuado para revertir la CID es la correcta reposición del volumen. Sin embargo, en la práctica diaria, se observan resultados favorables con el uso de heparina (dosis de 7,5 a 12 Ud/ kg/ hora). En la mayoría de los casos se logra la detección del proceso en 4 a 6 horas, apoyados con el resto de las medidas encaminadas a la extirpación del foco séptico. Es conocida la influencia de la sepsis en la disminución de las concentraciones de ATIII, de manera que se recomienda el empleo de ATIII para revertir la CID por este proceso infeccioso.
La muerte fetal intrauterina, acompañada de un estado de retención de 4 a 5 semanas establece el riesgo de CID en 40 % de las pacientes; sin embargo, es cada vez menos frecuente la aparición de estos trastornos asociados al feto muerto, teniendo en cuenta la conducta activa, dada por la inmediata interrupción del embarazo en la mayoría de las gestantes. La expulsión diferida espontánea en embarazos a término (de 75 - 85 % en las 2 - 3 últimas semanas) ha dejado de ser una práctica. La modificación que sufre la placenta y el feto mediante el proceso de maceración, facilita la entrada de sustancia tromboplástica a la circulación, lo que desencadena la CID. Excepcionalmente autores utilizan la heparina si existe síndrome de feto muerto y, de preferencia, si se diagnostica la fase de hipercoagulabilidad, en nuestra experiencia nunca la hemos empleado en esta indicación.
Mark y Zilliacus consideraron en 1948 el uso de heparina en pacientes con preclampsia lo cual no logró imponerse ante la regresión del cuadro en los pacientes con el tratamiento adecuado de la enfermedad de base.
Los criterios para la utilización de la heparina, en el embolismo del líquido amniótico cuando no está presente la coagulopatía, son controvertidos. Duff y Resneik no están de acuerdo en usarlo habitualmente; mientras que Weiner sugiere dosis de 3 000 _ 5 000 Ud por vía endovenosa y Cruikshank recomienda 10 000 Ud en el momento del diagnóstico de coagulopatía, seguidos de 5 000 Ud cada 6 horas.
Teniendo en cuenta las propiedades procoagulantes del líquido amniótico y la incidencia de este trastorno en 40_83 % de las pacientes, los autores de este artículo justifican el uso de esta terapia, en dosis de 5,000 Ud, por vía endovenosa, cada 4 horas.
Por el contrario, los que no están de acuerdo con el empleo de la heparina en el embolismo del líquido amniótico, aseguran que esta puede iniciar o agravar el sangrado y que no tiene efectos sobre los microtrombos ya formados; también se plantea que esta se inactiva en un pH menor de 7,2, por lo cual es improbable que pueda actuar en este tipo de paciente severamente acidótico. De los anteriores planteamientos se deduce la importancia de escoger el momento adecuado para su utilización.
Los antifibrinolíticos tienen poca participación en estos estados, teniendo en cuenta que la fibrinólisis es un fenómeno secundario a la CID y necesario para la recanalización del vaso y la reperfusión de órganos. Su utilización puede estar justificada cuando habiéndose utilizado heparina, aparece una actividad fibrinolítica exagerada. La sospecha se presenta cuando existe lisis espontánea del coagulo y se confirma déficit de factor V, VIII, fibrinógeno, tiempo de lisis de euglobina acortado, niveles de PDF aumentados y plaquetas normales básicamente esto último en la fibrinólisis primaria.
Las drogas más usadas internacionalmente son aprotinina y el ácido épsilon aminocaproico. La primera es más recomendada por su efecto inhibidor reversible de las proteasas séricas plasmina y los factores de la coagulación. La dosis recomendada es de 200 000UI por hora, en infusión intravenosa, con vida media de 40 minutos. Los riesgos trombogénicos son menores que los presentados con el uso de otros antifibrinolíticos en los que debe haberse utilizado heparina previamente; la segunda, debe usarse en dosis de 4 a 6 gramos, en infusión intravenosa lenta y después 1 gramo por hora, bloquea los sitios de unión del plasminógeno sobre el fibrinógeno y la fibrina.
De forma excepcional pudiera considerarse su uso en obstetricia en el embolismo del líquido amniótico y en el desprendimiento de la placenta normoinserta cuando se presentaran los criterios antes expuestos.
CONCLUSIONES
En el período grávido-puerperal, la coagulación sanguínea sufre cambios que se impone conocer para interpretar correctamente esos trastornos cuando se presentan en esta etapa, teniendo en cuenta las posibles complicaciones y el peligro para la vida que pueden presentarse. Asimismo, está demostrada la necesidad de diagnosticar la CID en la fase bioquímica de bajo grado, como premisa para lograr respuesta al tratamiento, así como también para reducir el daño de órganos y la mortalidad. Por otra parte, el conocimiento de las enfermedades que desencadenan la CID y la interpretación adecuada de los medios de diagnósticos, es fundamental para lograr su identificación temprana. Resulta elemental aplicar en estas pacientes el tratamiento expedito de la enfermedad de base para lograr la resolución del cuadro y eliminar las complicaciones.
REFERENCIAS BIBLIOGRAFICAS
1. Almagro Vázquez D. La hemostasia en las complicaciones obstétricas. La Habana: ECIMED; 1997.
2. Altman R, Rouvier J, Reussi R. Coagulación intravascular diseminada. Rev Iberouner Tromh Hemostasia. 1994;7: 272-92.
3. Nápoles Méndez D, Couto Núñez D. Experiencia de 11 años en enfermedad tromboembólica venosa en el período grávido puerperal. Rev Cubana Obstet Ginecol [Internet]. 2011 [citado 12 Ago 2011]; 37(3). Disponible en: http://bvs.sld.cu/revistas/gin/vol37_3_11/gin02311.htm
4. The American College of Obstetricians and Gynecologists. Thromboembolism in pregnancy. Pract Bull. 2011; 118(3):718-29.
5. Roca Goderich R, Smith Smith V, Paz Precilla E, Losada Gómez J, Serret Rodríguez B, Llamos Sierra N, et al. Hemostasia. En: Temas de medicina interna. 4 ed. La Habana: ECIMED; 2002. p. 385-9.
6. Caballero López A. Trastornos de la coagulación en el paciente grave. En: Terapia intensiva. La Habana: ECIMED; 2008. p. 1205-15.
7. Vázquez Cabrera J. Trastorno de la hemostasia durante el embarazo, parto y puerperio. En: Embarazo, parto y puerperio. Principales complicaciones. La Habana: ECIMED; 2009. p. 136-56.
8. Fernández Mirabal J. La coagulación de la sangre. La Habana: Editorial Científico Técnica; 1975.
9. Nápoles Méndez D, Couto Núñez D. Enfermedad tromboembólica venosa en el embarazo y puerperio. Parte 1. Enfoque de riesgo y diagnóstico. MEDISAN [Internet]. 2011 [22 Ene 2012]; 15(10). Disponible en: http://bvs.sld.cu/revistas/san/vol_15_10_11/san121011.htm
10. Vázquez Cabrera J. Embolismo del líquido amniótico. En: Embarazo, parto y puerperio. Principales complicaciones. La Habana: ECIMED; 2009. p. 158-62.
11. Caballero López A. Embolia del líquido amniótico. En: Terapia intensiva. La Habana: ECIMED; 2008. p. 1313_20.
12. Martí Carvajal A, Comunián Carasco G, Peña Martí G. Haematological interventions for heating disseminated intravascular coagulation during pregnancy and pospartum. Cochrane Database Systematic Reviews [Internet]. 2011 [22 Ene 2012]; Issue 3. Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/14651858.CD008577.pub2/pdf
13. Saba HI, Morelli GA. The pathogenesis and management of disseminatied intravascular coagulation. Clin Adv Hematol Oncol. 2006; 4:919_ 26.
14. Montagnana M, Franchi M, Danese E, Gotsch F, Guidi GC. Disseminated intravascular coagulation in obstetric and gynecologic disorders. Seminars in Thrombosis Hemostasis. 2010; 36:404_18.
15. Levi M. Disseminated intravascular coagulation (CID) in pregnancy and the peri-partum period. Thromb Res. 2009; 123(2):63_4.
16. Iglesias Almanza NR, Guirola de la Pana J, Pérez Assef H, Fernández Gutierrez R, Herrera Collado R, Morales Martinez H. Trastornos de la coagulación en el embarazo. MEDICIEGO [Internet]. 1999 [citado 20 Dic 2010]; 2(1). Disponible en: http://bvs.sld.cu/revistas/mciego/vol2_01_96/a8_v2_0196.html
17. Roca Goderich R, Smith Smith V, Paz Precilla E, Losada Gómez J, Serret Rodríguez B, Llamos Sierra N, et al. Coagulación intravascular diseminada. En: Temas de medicina interna. 4 ed. La Habana: ECIMED; 2002. p. 402-5.
18. Almaguer Almaguer JA, López Artze O, Granado Martínez O, Hernández Núñez J, Valdés Yong M, Moreno Autié M. Embolismo del líquido amniótico vs hipertensión pulmonar primaria. Presentación de un caso. Rev Cubana Obstet Ginecol [Internet]. 2007 [citado 20 Dic 2010]; 33(3). Disponible en: http://bvs.sld.cu/revistas/gin/vol33_3_07/gin04307.html
19. Coronado Mestre R, Álvarez Pineda AB, Yero Castañeda M, Matos Ross O. Aborto séptico: síndrome de Mondor. Rev Cubana Med Milit [Internet]. 2006 [citado 20 Dic 2010]; 35(4). Disponible en: http://bvs.sld.cu/revistas/mil/vol35_4_06/mil09406.htm
20. Orizondo Ansola R. Novedades y controversias en relación con la preeclampsia/ eclampsia. Rev Cubana Med [Internet]. 2007 [citado 20 Dic 2010];46(2). http://scielo.sld.cu/scielo.php?pid=S0034-75232007000200009&script=sci_arttext
21. Cortina Rosalkes LD, López de RRowx MR. Terapia transfusional en la hemorragia obstétrica mayor. Síndrome de transfusión masiva. Rev Cubana Obstet Ginecol [Internet]. 2007[citado 20 Dic 2010]; 33(3). Disponible en: http://bvs.sld.cu/revistas/gin/vol33_3_07/gin06307.html
22. Bruhn A, Pairumanu R Hernández G. Manejo del paciente en shock séptico. Rev Med Clin Condes. 2011; 22(3):293-301.
23. Guinn D, Abel D, Tomlinson M. Early goal directed therapy for sepsis during pregnancy. Obstet Gynecol Clin Nam. 2007; 34:459-79.
24. Martínez Murillo C, Quintana González S. Farmacología de los antitrombóticos. Gac Med Mex. 2007; 143(1):25-8.
25. Monreal Busch M. Indicaciones actuales del tratamiento con heparina. Rev Clin Esp. 2006;(2):98-9.
26. Walker MC, Ferguson SE, Allen VM. Heparina para las mujeres embarazadas con trombofilia adquirida o heredada. Biblioteca Cochrane Plus. [Internet]. 2008 [citado 20 Dic 2010]; 2. Disponible en: http://www.update-software.com/pdf/CD003580.pdf
27. Heineger AI. Nuevos anticoagulantes orales. Taller de anticoagulación, Málaga 2009. [Internet]. [citado 20 Dic 2010]. Disponible en:http://www.apam-malaga.org/uploads/5/6/9/3/569318/nuevos_anticoagulantes_orales.pdf
28. González Popoca M. Trombosis y embarazo. Rev Hemo Trombo. 2008; 2(2-4): 121-7.
29. Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2008; 133(suppl 6):160-98.
30. Kitchens CS. Thrombocytopenia and thrombosis in disseminated intravascular coagulation (DIC). Hematology Am Soc Hematol Educ Program. 2009:240-6.
31. Mazzuccomi MG, de Sanctis V, Dragoni F, Chistolini A, Dinucei G, Petrelli V, et al. La coagulación intravascular diseminada en obstetricia y ginecología (CID). [Internet]. [citado 20 Dic 2010]. Disponible en: http://www.sepeap.org/archivos/libros/HEMATOLOGIA/coagulacion/11.pdf
32. Tasnim S. Disseminated intravascular coagulation in obstetrics. Orion Medical Journal. 2004; 18:182–4.
33. Thachill J, Toch CH. Disseminated intravascular coagulation in obstetric disorders and its acute haematological management. Blood Rev. 2009; 23(4):167-76.
34. Malvino E. Uso de factor VII activado recombinante en obstetricia. Obstetricia crítica. [Internet]. 2006 [citado 20 Dic 2010]. Disponible en: http://www.obstetriciacritica.com.ar/doc/nota1014.pdf
35. Nápoles Méndez D, Couto Núñez D. Enfermedad tromboembólica venosa en el embarazo y puerperio. Parte 2. Prevención y tratamiento. MEDISAN. [Internet]. 2011 [22 Ene 2012]; 15(11). Disponible en: http://bvs.sld.cu/revistas/san/vol_15_11_11/san101111.htm
36. Willians J, Mozurkewich E, Chilimigras J, Ven C. Critical care in obstetrics: pregnancy–specific conditions. Best Practice & Research. Clinical obstetrics & gynaecology. 2008; 22: 825-46.
37. Attila P. Thromboembolia-antithromboticus. Kezlebes terhessegben. Orw Hetil. 2011; 152:815-21.
38. James AH. Pregnancy-associated thrombosis. Hematoloy [Internet]. 2009 [citado 25 May 2011].Disponible en: http://asheducationbook.hematologylibrary.org/cgi/content/full/2009/1/277
39. Rodger M. Evidence base for the management of venous thromboembolism in pregnancy. Haematology [Internet]. 2010 [citado 25 May 2011]. Disponible en: http://asheducationbook.hematologylibrary.org/cgi/content/full/2010/1/173
Recibido:14 de octubre de 2011
Aprobado: 26 de enero de 2012
Danilo Nápoles Méndez. Hospital Provincial Ginecoobstétrico "Mariana Grajales Coello", avenida Victoriano Garzón, Santiago de Cuba, Cuba. Correo electrónico: danilon@medired.scu.sld.cu

