










 Custom services
Custom services Portuguese (pdf)
Portuguese (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntrodução
A Amelogênese imperfeita (AI) é uma condição de desenvolvimento genômica que se caracteriza por hipomineralização ou hipoplasia com descoloração, sensibilidade e fragilidade por meio de defeitos variados na matriz do esmalte dentário.1 A prevalência com relação ao acometimento da AI varia entre as populações e os métodos de diagnóstico, de 1:700 até 1:14000,2 tendo em vista que essa condição afeta não apenas a aparência clínica, mas também a estrutura dental de praticamente todos os dentes, exibindo características heterogêneas entre os pacientes envolvidos.3
A etiologia é incerta e vem sendo esclarecida ao longo do tempo, sendo ligada a um padrão de herança variável: ligada ao cromossomo X, autossômica, dominante ou recessiva,4 estando relacionada a uma série de genes como AMELX, ENAM, KLK4, MMP20, FAM83 e WDR72.4,5 A diversidade de fenótipos encontrados associados a AI é reflexo do momento em que ocorre a falha ou ruptura da normalidade do processo da amelogênese,1 que pode resultar nas seguintes variantes clínicas: hipoplásica, hipocalcificada, hipomaturada e hipoplásica-hipomaturada com taurodontismo.1
A variante hipoplásica é a de maior prevalência entre os 4 tipos clínicos, correspondendo entre 60 % e 73 % dos casos, com defeito ocorrendo durante a formação da matriz do esmalte.6 Na AI hipocalcificada a anormalidade está nas etapas de mineralização do esmalte, representando 7 % dos casos totais já descritos.6 A hipomaturada apresenta problemas relacionados a defeitos na maturação do esmalte, representando de 20 % a 40 % dos casos registrados.6 Existe ainda um quarto tipo de AI, a hipoplásica-hipomaturada com taurodontismo, que atinge uma menor parcela dos portadores dessa condição.6,7
Além dos distúrbios locais, a AI também pode estar associada a complicações sistêmicas, conhecida como síndrome da amelogênese imperfeita e nefrocalcinose,7 a síndrome de Jalili8 e a fibromatose gengival.9
Discutir as alterações clínicas e a importância de um diagnóstico correto para direcionar a melhor conduta deve ser o foco principal do tratamento. Sendo assim, este trabalho pretende relatar dois casos de amelogênese imperfeita do tipo hipoplásica entre membros de uma mesma família, correlacionando-os.
Apresentação do caso
Caso 1
Paciente do sexo masculino, 16 anos, compareceu à clínica de Propedêutica Estomatológica da Universidade Federal de Campina Grande, campus de Patos-PB, queixando-se da aparência estética dos elementos dentais. Segundo a mãe do paciente assemelhavam-se a “dentes de rato, pequenos e finos”, além do relato de que também havia falta de alguns dentes.
Durante a anamnese não foram relatados dados significativos relacionados a alguma alteração sistêmica, deficiência nutricional ou tratamento anterior com drogas que justificasse as características relatadas inicialmente pelo paciente.
No exame físico extraoral o paciente apresentou protrusão mandibular e maxila levemente retrusa, associada à diminuição da largura do terço inferior da face e perfil côncavo (Fig. 1, A, B, C).
No exame físico intraoral, o paciente foi classificado como classe III de Angle com mordida cruzada posterior. Percebeu-se a presença de fossetas e sulcos distribuídos aleatoriamente em todas as faces de todos os elementos dentais presentes, com severidade clínica variável. Essas depressões além de exibirem coloração marrom-amarelada, possuíam ainda no espaço entre elas um esmalte com dureza, textura e coloração semelhantes à normalidade (Fig. 1, D, E, F).
Os elementos 11 e 21 já haviam sido restaurados com resina composta em outro serviço, entretanto nos dentes posteriores, local de maior incômodo relatado pelo paciente por conta da sensibilidade dental, notou-se grande desgaste da superfície oclusal com exposição dentinária, devido à fragilidade do esmalte presente. Notava-se ainda a falta de elementos dentais (16, 17, 18, 27, 28, 37, 38, 47 e 48), visíveis pela presença de espaços edêntulos, nas arcadas superior e inferior.
No exame radiográfico panorâmico constatou-se a presença de todos os elementos dentais não visíveis no exame intraoral. O esmalte apesar de bastante delgado, radiograficamente apresentava-se com radiopacidade semelhante à normalidade, o que ainda permitiu diferenciá-lo em contraste com a dentina. Todos os elementos apresentavam ainda câmara pulpar em dimensões de normalidade, sem nenhuma alteração significativa de condutos ou alargamento da câmara. Foi possível evidenciar ainda uma lesão periapical no primeiro molar esquerdo inferior com reabsorção da raiz mesial (Fig. 1, G).
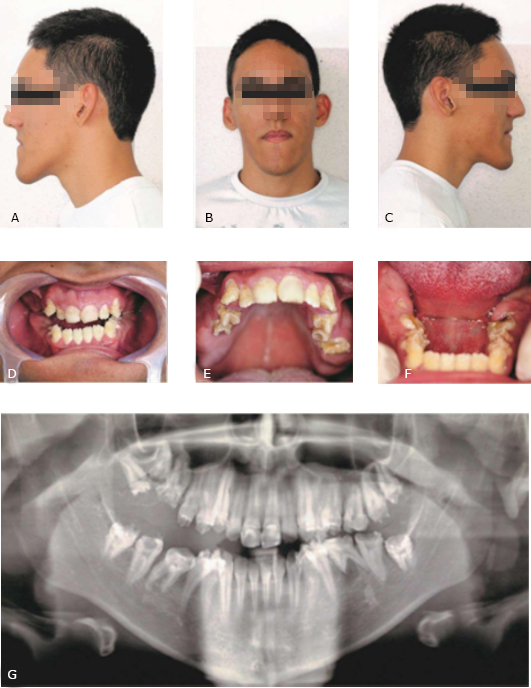
Caso 2
Paciente do sexo masculino, 13 anos, compareceu ao serviço por recomendação do tio (caso anteriormente citado) que apresentava as mesmas características. Queixava-se no seu atendimento inicial de sensibilidade em alguns elementos dentários, atrelado à descoloração dental, somando-se a descontentamento estético: “acho os dentes da frente feio e esses de trás são muito sensíveis”. Não foi relacionada nenhuma alteração sistêmica digna de nota.
No exame extraoral, o paciente apresentou pequena protrusão maxilar e notável retrusão mandibular, com perfil convexo e a presença de um selamento labial forçado, causado através de uma mordida aberta anterior, visível no exame intraoral (Fig. 2, A, B, C).
No exame intraoral o paciente foi classificado como classe I de Angle, apresentando aspectos relacionados a características oclusais posteriores próximos da normalidade, contudo, havia a presença de uma mordida aberta anterior. Percebeu-se a presença de fossetas e sulcos pigmentados marrom-amarelado em todas as faces dos elementos dentais, distribuídos aleatoriamente e circundados por regiões de esmalte alternado com esmalte de dureza, textura e coloração semelhantes à normalidade, tanto na arcada superior, quanto inferior, anterior e posterior (Fig. 2, D, E, F).
Foi possível observar a falta de alguns elementos dentais (17, 27, 37, sendo o 47 parcialmente erupcionado, com 18 e 28 em processo de formação, visível através do exame radiográfico) e grande desgaste oclusal nos dentes posteriores. O paciente apresentava ainda alguns elementos já restaurados com resina composta e outros com lesões de cárie e acúmulo de biofilme dentro das fossetas e sulcos dos dentes afetados com AI.
Com relação ao exame radiográfico panorâmico, observou-se um esmalte bastante delgado, porém com radiopacidade e contraste em normalidade, o que permitiu a diferenciação da dentina. Também não se evidenciou a presença de qualquer alteração na câmara ou cornos pulpares, como alargamentos, estreitamentos ou calcificações. Todos os elementos não visíveis no exame intraoral, foram percebidos através da análise radiográfica. Assim como o caso 1, este paciente também apresentava lesão periapical no elemento dentário 36 (Fig. 2, G).
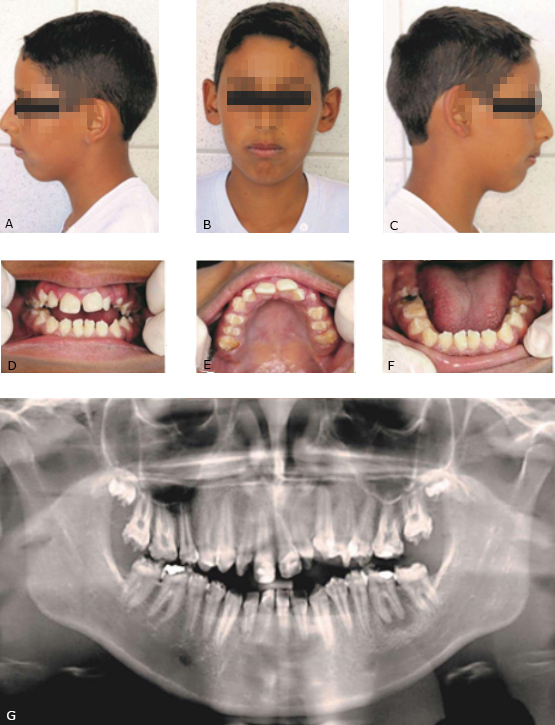
Fig. 2- Aspecto extraoral, intraoral, clínico e radiográfico. A, B, C: Paciente com padrão esquelético de retrusão mandibular; D, E, F: Observar a presença generalizada da AI, com características clínicas exuberantes de sulcos e fossetas pigmentadas; G: Radiografia panorâmica mostrando elementos com radiopacidade esmalte/dentina contrastante.
Os pacientes relataram que em sua família outros indivíduos tinham histórias similares de acometimento dentário. A mãe do caso 1 relatou fazer uso de prótese dental total superior e inferior, pois havia perdido todos os elementos devido à condição dentária semelhante à do filho, o que a levou a um sério desgaste dental com perda de dimensão vertical e posterior remoção de todos os restos radiculares. Após coleta de informações inerentes a outros parentes afetados e análise da genealogia em heredograma (Fig. 3), sugerimos que nesta família a AI apresenta-se com caráter hereditário autossômico dominante, pois em todas as gerações que se pode estudar pelo menos um indivíduo tinha a doença independentemente do sexo.
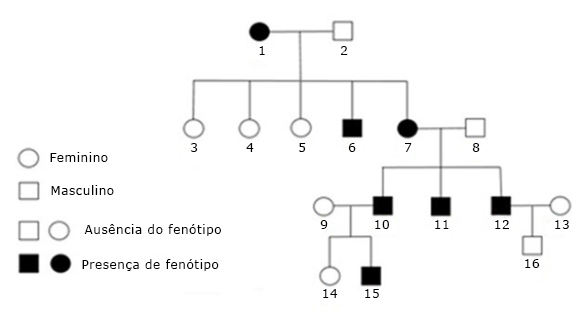
Fig. 3- Heredograma traçado a partir da história familiar. Percebe-se que a AI afetava todas as gerações, sem predileção por sexo, fortalecendo a hipótese de amelogênese imperfeita com caráter autossômico dominante. Os pacientes relatados estão identificados pelos números 11 e 15.
Diante disso, e com ênfase nas características clínicas dos subtipos presentes na AI, os dois pacientes foram classificados em amelogênese imperfeita do tipo hipoplásica (AIH). Os pacientes foram encaminhados para o tratamento multidisciplinar envolvendo diversas especialidades odontológicas como periodontia, endodontia, dentística, prótese, ortodontia e cirurgia.
Discussão
A AI é uma alteração genética de características bastante heterogêneas, incluindo dentes propensos a desgastes ou desintegração rápida, problemas de hipersensibilidade, alteração da normalidade de oclusão, alterações periodontais e outras condições que necessitam de tratamentos extensivos restauradores e reparadores.10
Esta condição apresenta heterogeneidade na sua etiologia, estando associada a mutações em muitos genes que codificam proteínas do esmalte. A variedade da expressão genética afeta não apenas a quantidade, mas também a qualidade do esmalte presente em ambas às dentições,11 causando também falhas no processo de erupção normal dos elementos dentários, sendo até seis vezes maior a chance do que na população normal. Na história clínica, os pacientes relataram que apresentaram essas manchas tanto na dentição decídua, quanto na permanente, assim como demora da erupção de vários elementos dentais.
O esmalte dentário é feito de cristais de hidroxiapatita altamente organizados, ocupando 95% do volume da matriz. O padrão diversificado visível na malformação do esmalte, vai refletir o momento da falha da normalidade do processo, produzindo uma série de defeitos fenotípicos.1 Estes defeitos se dividem em: hipoplásica, hipocalcificada, hipomaturada e hipoplásica - hipomaturada com taurodontismo.1
A variante da AI hipoplásica representa a maioria de todos os casos relatados (60 % a 73 %).3 É descrita e caracterizada por inúmeros estudos como defeitos na fase de secreção da matriz do esmalte, resultando no alongamento insuficiente de cristais de esmalte, tendo a característica clínica de esmalte delgado, com sulcos ou fossetas locais ou desorganizadamente generalizados, com coloração semelhante ao marrom-amarelada, perda patológica do esmalte devido a fragilidade e o contato áspero durante o uso, porém com características radiográficas de radiodensidade normais e contrastante com a dentina.1,3,6,12 Os casos expostos apresentavam as características acima descritas e por isso o diagnóstico foi de AI do tipo hipoplásica generalizada.
Para o diagnóstico deve-se levar em relação a história familiar associado às características clínicas e radiográficas para determinar a variante de AI, assim como a exclusão de outras condições que afetem o esmalte dentário, como coloração por tetraciclina, dentinogênese imperfeita ou ainda hipomineralização molar-incisivo (MIH).1 A MIH é a que apresenta maior semelhança com a AIH, porém afeta principalmente os 2/3 oclusais da coroa de molares e incisivos, diferindo do aspecto generalizado da AI.13 Na dentinogênese imperfeita é comum além dos danos no processo normal de formação da dentina a presença de alterações pulpares.14 Os casos relatados apresentavam fossetas e sulcos em todas as faces de todos os elementos dentais, porém sem alterações pulpares. Este padrão foi fundamental para descartarmos outras hipóteses diagnósticas.
A presença de mordida aberta anterior, colapso oclusal posterior e aumento do terço ântero-inferior da face, são frequentemente encontradas nesses pacientes, devido disposição incorreta da língua, ocasionado pela sensibilidade dentária ou pelo colapso posterior relacionado ao mal posicionamento destes elementos, gerando ainda condições esqueléticas relacionadas a classe III e II de Angle, com características semelhantes entre os membros da mesma família.15
A junção da AI com condições sistêmicas, como por exemplo a nefrocalcinose, apresenta poucos casos relatados, porém com significativa morbidade, por alterações da função renal,16 conectando-se a nefrocalcinose principalmente ao fator recessivo.17 Os casos relatados através da história familiar, pode-se evidenciar que todas as gerações tinham casos de AI, sendo sugerida herança dominante.
Com relação à síndrome de Jalili, esta é tratada como uma condição de mutação do gene CNNM4 que afeta o fluxo de íons magnésio, gerando um distúrbio de degeneração dos fotorreceptores da retina culminando com deficiência visual progressiva, associando-se a hipomineralização do esmalte, caracterizando-o como amelogênese imperfeita do tipo hipocalcificada.8 Os pacientes retratados apresentaram a variante hipoplásica da AI, além disso, não esteve presente em nenhum dos avaliados a presença de doença oftálmica pregressa ou presente, o que pode ser sugestivo da não presença desta síndrome.
Outra condição sistêmica relevante que pode se apresentar entre os pacientes com AI, é a presença da fibromatose gengival, que através de um estudo de caso num paciente do Marrocos, foi identificado uma mutação no gene FAM20A, também responsável pela AI, a qual apresentou as duas condições em associação, sendo ligado a um padrão recessivo da AIH.9 Apesar dos dois pacientes deste caso apresentarem uma camada de gengiva espessa recobrindo alguns elementos dentais, esta não foi caracterizada como fibromatose gengival, além disso, pode-se excluir a hipótese desta associação devido ao fato da AIH ter se apresentado em todos as gerações da família, o que pode ser indicativo de uma condição dominante.
O tratamento de pacientes com esse tipo de condição se dá de maneira individualizada e multidisciplinar, abordando as inúmeras condições apresentadas, não só de forma estética e funcional, mas também contribuindo de forma psicologicamente positiva, sendo de forma contínua e de acompanhamento constante.2
Conclui-se assim que além da multidisciplinaridade para o tratamento clínica, também vale ressaltar a importância do aconselhamento genético para os indivíduos acometidos e, assim, prever tratamento e prognóstico destes.

