










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Farmacia
On-line version ISSN 1561-2988
Rev Cubana Farm vol.36 no.1 Ciudad de la Habana Jan.-Apr. 2002
Centro de Investigación y Desarrollo de Medicamentos
Validación de los métodos analíticos para la identificación y cuantificación del dextrometorfano jarabe
Alfredo Fernández Serret,1 Yeni Aguilera Cabrera,2 Iván Morales Lacarrere3 y Esther Alonso Jiménez4
Resumen
Para realizar el control de la calidad del dextrometorfano jarabe, se aplicó la técnica descrita en la USP 23, y se comprobó la fiabilidad de esta mediante la evaluación de la precisión, exactitud, linealidad y especificidad. Se incluyen además, la determinación de las características organolépticas, pH y contenido por frasco como ensayos propios de medicamentos líquidos de administración oral o de uso externo.
DeCS: DEXTROMETORFANO/análisis; DEXTROMETORFANO/farmacología; CONTROL DE CALIDAD; CROMATOGRAFIA LIQUIDA DE ALTA PRESION; CALIDAD DE LOS MEDICAMENTOS; TOS/quimioterapia; QUIMICA FARMACEUTICA.
La validación es el proceso establecido para la obtención de pruebas documentadas y demostrativas de que un método de análisis es lo suficiente fiable y reproducible para producir el resultado previsto dentro de intervalos definidos (Validación de métodos analíticos. Monografía. Sección catalana de la AEFI. Comisión de normas de buena fabricación y control de calidad. Sept., 1989:1-94).
La validación proporciona un alto grado de confianza y seguridad del método analítico y se realiza con carácter obligatorio cuando se desarrolla un nuevo procedimiento, ya que permite asegurar que el método propuesto hace lo que tiene que hacer.1
Es necesario señalar que los métodos descritos en farmacopeas u otros textos oficiales se consideran validados, aunque debe aclararse que ellos se refieren solamente a métodos generales y a materias primas. Estos no precisan de validación, aunque deben ser comprobados antes de su utilización rutinaria con la verificación de la idoneidad en las condiciones de laboratorio. Sin embargo, para el caso de las formas farmacéuticas terminadas, que pueden variar su composición cualitativa o cuantitativa según el fabricante, habrá que proceder en cada caso a la validación del método analítico. Para demostrar la aplicabilidad de estos métodos que no tienen en cuenta todos los posibles excipientes de una forma farmacéutica, será necesario, por lo menos, efectuar una validación abreviada que evalúe que la especificidad, exactitud y precisión son adecuadas (Validación de métodos analíticos. Monografía. Sección catalana de la AEFI. Comisión de normas de buena fabricación y control de calidad. Sept., 1989:1-94).
El presente trabajo tuvo como objetivo la validación del método analítico reportado en la USP 232 para el control de la calidad del dextrometorfano jarabe, medicamento antitusivo de gran eficacia empleado ampliamente para combatir los accesos de tos.3
Métodos
- Para el desarrollo de este estudio se empleó como materia prima el bromhidrato de dextrometorfano (C18H25NO·HBr·H2O), perteneciente al lote 910156 del fabricante La Roche, y muestras de los lotes 31, 32 y 33 para evaluar las especificaciones de calidad del dextrometorfano jarabe.
Métodos analíticos para el control de calidad
Se empleó la técnica analítica descrita en la monografía para el control de la calidad del jarabe de dextrometorfano según la USP 232 que incluye los ensayos siguientes:
Identificación. Se basa en la comprobación de la propiedad dextrorrotatoria de la solución de bromhidrato de dextrome-torfano obtenida después de un tratamiento de extracción al jarabe. Las mediciones se realizaron en un polarímetro Carl Zeiss. Este ensayo se fundamenta en una reacción química de coloración la cual permite identificar compuestos que contienen el grupo O- alquil unido a un anillo bencénico o a un anillo en una estructura policíclica que contiene un anillo bencénico.
Cuantificación. Se basa en la separación selectiva del bromhidrato de dextrometorfano, mediante la cromatografía líquida de alta resolución de fase reversa por formación de par iónico y la cuantificación de este, frente a una muestra de referencia que emplea el método de comparación absoluta o patrón externo. Se empleó un cromatógrafo líquido equipado con un detector UV, y se establecieron las condiciones operacionales siguientes:
- Flujo de la fase móvil: 1,0 mL/min.
- Detector UV a longitud de onda: 280 nm.
- Sensibilidad del detector: 0,08 AUFS.
- Volumen inyectado: 20 µL.
Se incluyen además, la determinación de las características organolépticas, pH y contenido por frasco como ensayos propios para el control de la calidad de los medicamentos líquidos de administración oral o de uso externo.4
Validación de los métodos de identificación del principio activo
Especificidad. Se comprobó la especificidad de los métodos de identificación, mediante la aplicación de las metodologías descritas en la monografía a una muestra placebo de la formulación.
Validación del método de cuantificación del principio activo
Linealidad del sistema. Para el estudio de la linealidad del sistema se preparó una curva de calibración en un intervalo de concentración desde 24,0 hasta 192,0 µg/mL (20-160 % de la cantidad teórica declarada), que incluyó 7 concentraciones diferentes. El análisis se realizó por triplicado.
Precisión del método. En el caso de la repetibilidad, este estudio se realizó por quintuplicado a una muestra de con-centración única que representó el 100,0 % de la cantidad teórica declarada; mientras que para la reproducibilidad, se analizó con la misma muestra homogénea de concentración única, y se efectuaron las valoraciones por 2 analistas en 2 d diferentes. Para su evaluación se realizó un análisis de varianza.
Exactitud, linealidad del método e intervalo. Para el estudio de la exactitud se empleó el método de recuperación, me-diante la preparación de muestras con diferentes niveles de bromhidrato de dextrometorfano, que representan el 80, 90, 100, 110 y 120 % de la concentración teórica del principio activo en el jarabe, según la formulación propuesta. Simultáneamente, se analizaron la linealidad del método y el intervalo. El análisis se realizó por triplicado.
Especificidad. Con el objetivo de evaluar la especificidad del método cromatográfico, se sometió el placebo de la formulación seleccionada y el medicamento a hidrólisis ácida con ácido clorhídrico 3 M, alcalina con hidróxido de sodio 3 M y oxidación con peróxido de hidrógeno a una temperatura de 100 °C por 3 h. Además, el placebo fue sometido a calentamiento a 70 °C por 30 d.
Todos los resultados obtenidos fueron evaluados estadísticamente con la ayuda del programa Origin Versión 5.0 para Windows.
Resultados
Validación del método de identificación del principio activo
La solución obtenida al someter el placebo al proceso de extracción no presentó actividad óptica. De igual forma no se desarrolló coloración alguna indicativa de la presencia de este tipo de compuesto en la muestra de placebo ensayada.
Validación del método de cuantificación del principio activo
Linealidad del sistema. La curva de calibración a 280 nm resultó ser lineal en el intervalo de concentraciones comprendidos entre 24,0 y 192,0 µg/mL. Al aplicar el método de los mínimos cuadrados a los resultados registrados se obtuvo la ecuación de la recta que se expresó según y = 11 019,41 x – 2 909,67. El coeficiente de correlación lineal fue de 0,9998. Al aplicar la prueba de linealidad se obtuvo un valor de desviación estándar relativa del 0,22 % y al aplicar la prueba de proporcionalidad los límites de confianza del término independiente incluyen el cero.
Precisión del método. En la tabla 1 se exponen los resultados correspondientes a este estudio. Para el caso específico de la repetibilidad, los coeficientes de variación fueron menores que 1,5 % y el coeficiente de variación total menor que 2 %. Las F experimentales de Fisher resultaron ser menores que las tabuladas.
Tabla 1. Estudio de la precisión del método. Repetibilidad y reproducibilidad
| Analista | Día 1. Repetibilidad | Día 2. Repetibilidad | Reproducibilidad |
| Analista 1 | 1. 104,2 % | 6. 103,7 % | |
| 2. 103,9 % | 7. 104,4 % | ||
| 3. 103,8 % | 8. 104,1 % | Xmedia= 104,1 % | |
| 4. 104,0 % | 9. 103,1 % | ||
| 5. 104,6 % | 10. 104,3 % | s = 0,39 | |
| Xmedia = 104,1 % | Xmedia = 103,9 % | ||
| CV = 0,30 % | CV = 0,51 % | ||
| Analista 2 | 11. 104,4 % | 16. 104,2 % | n = 20 |
| 12. 104,0 % | 17. 103,8 % | ||
| 13. 104,5 % | 18. 104,0 % | ||
| 14. 104,0 % | 19. 103,9 % | CV= 0,38 % | |
| 15. 104,7 % | 20. 104,8 % | ||
| Xmedia = 104,2 % | Xmedia = 104,1 % | ||
| CV= 0,30 % | CV = 0,38 % |
Intervalo de confianza = 104,1 ± 0,2
Límites de aceptación: Repetibilidad CV ± 1,5 %
Reproducibilidad CV £ 2,0 %
| Fuente de | Grados de | Suma de | Medias de | ||
| variación | libertad (gl) | cuadrados | cuadrados | Fcal | Ftab |
| Analista | 1 | 0,242 | 0,242 | Fa | Fgla/Fgld |
| (a) | 2,9876 | 38,51 | |||
| Día | 2 | 0,162 | 0,081 | Fd | Fgld/Fgle |
| (d) | 0,5087 | 4,69 | |||
| Error | 16 | 2,548 | 0,1592 | ||
| (e) |
F = F de Fisher; gla = grados de libertad para los analistas;
gld = grados de libertad para los días;
gle = grados de libertad del error. Fa < Fgla/gld;
Fd < Fgld/gle.
Exactitud, linealidad del método e intervalo
Los resultados obtenidos en el estudio de la linealidad del método se muestran en la tabla 2, donde la pendiente de la curva de recuperación presentó un valor de 0,9972. La curva de recuperación mostró un comportamiento lineal dado por la ecuación de la línea recta y el coeficiente de correlación de 0,9993. La recuperación media fue del 98,7 %. Al aplicar la prueba de linealidad se obtuvo un valor de desviación estándar relativa igual al 1,0 % y el intervalo de confianza del término independiente incluyó el cero.
Tabla 2. Exactitud del método. Curva de recuperación
| mg | % de | ||
| mg añadidos | recuperados | recuperación | Precisión |
| 96,0 | 94,5 | 98,4 | Xmedia = 99,0 % |
| 95,4 | 99,4 | S2 = 0,30 | |
| 95,3 | 99,3 | CV = 0,56 % | |
| 108,0 | 105,6 | 97,8 | Xmedia = 98,1 % |
| 106,8 | 98,9 | S2 = 0,49 | |
| 105,4 | 97,6 | CV= 0,71 % | |
| 120,0 | 118,6 | 98,8 | Xmedia = 98,5 % |
| 118,2 | 98,5 | S2 = 0,062 | |
| 118,0 | 98,3 | CV= 0,52 % | |
| 132,0 | 130,7 | 99,0 | Xmedia = 99,1 % |
| 130,3 | 98,7 | S2 = 0,26 | |
| 131,6 | 99,7 | CV = 0,52 % | |
| 144,0 | 142,5 | 98,9 | Xmedia = 98,9 % |
| 141,9 | 98,5 | S2 = 0,123 | |
| 142,9 | 99,2 | CV = 0,36 % |
Límites de aceptación: Rmedia = 98,7 %
R media 98,0 – 102,0 % CV = 0,58 %
CV£ 2,0 % Ecuación de la curva de recuperación:
y = 0,9972 x – 1,1533.
r = 0,9993.
Especificidad. En la figura 1 se presentan los cromatogramas de las muestras placebos sometidas a condiciones drásticas. En todos los casos se observa la aparición de picos secundarios que eluyen a tiempos de retención pequeños, los cuales modifican el cromatograma de la muestra placebo.
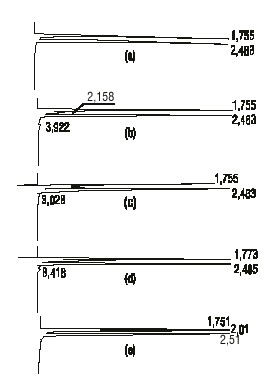
FIG. 1. Cromatogramas de las muestras placebos sometidas a condiciones drásticas: a) placebo sin degradar, b) placebo degradado a 70 °C, c) hidrólisis ácida del placebo, d) hidrólisis básica del placebo y e) oxidación con peróxido de hidrógeno.
Los cromatogramas de las muestras de medicamento sometidas a hidrólisis ácida, básica y oxidación con peróxido de hidrógeno se presentan en la figura 2.

FIG. 2. Cromatogramas de las muestras del medicamento sometidas a condiciones drásticas: a) medicamento sin degradar, b) hidrólisis ácida de la muestra, c) hidrólisis básica de la muestra y d) oxidación con peróxido de hidrógeno.
No se observó una hidrólisis apreciable en medio ácido ya que al término de 3 h con tratamiento con ácido clorhídrico (b) se obtuvo una concentración del 97,8 % de principio activo. Sin embargo, la hidrólisis en medio básico (c) mostró una degradación más acentuada, para una concentración del 76,0 % de dextrometorfano. De igual forma, se obtuvo la degradación del medicamento en medio oxidante (d), con el 66,0 % de este después de sometida la muestra al tratamiento.
Los resultados del control de la calidad de los lotes estudiados se presentan en la tabla 3.
Tabla 3. Resultados analíticos del control de la calidad
| Ensayos | Lote 31 | Lote 32 | Lote 33 | Especificaciones |
| Características | Responde | Responde | Responde | Líquido ligeramente |
| organolépticas | siruposo, incoloro, olor | |||
| característico y sabor | ||||
| dulce-amargo | ||||
| Identificación | A) Responde | A) Responde | A) Responde | A)Solución |
| dextrorrotatoria | ||||
| B) Responde | B) Responde | B) Responde | B) Coloración amarilla | |
| que torna a rojo. | ||||
| Contenido del | 119,0 mL | 118,0 mL | 119,0 mL | No menor de 118,0 mL |
| frasco | ||||
| pH | 3,88 | 3,88 | 3,90 | 3,8-4,20 |
| Valoración | 102,3 % | 103,1 % | 103,4 % | 95,0-105,0 % |
Discusión
Los ensayos realizados para el estudio de especificidad en la muestra placebo, comprobaron que los métodos descritos resultaron ser específicos. Para ambos casos, ninguno de los excipientes presentes en la formulación interfieren en los ensayos de identificación propuestos en la monografía.
Se comprobó el cumplimiento de la linealidad del sistema en el intervalo de concentraciones estudiado por el elevado valor del coeficiente de correlación alcanzado. El valor de la desviación estándar relativa resultó adecuado si se tiene en cuenta que el valor aceptable debe ser menor o igual que 2 %. De igual forma el intercepto de la curva no fue significativo.
Los coeficientes de variación resultaron ser menores que 1,5 %, lo cual demuestra la buena repetibilidad del método de cuantificación. No se observaron diferencias significativas entre las medias de ambos analistas, ya que Fa<Fgla)gld, y el método analítico fue reproducible en distintos días por un mismo analista. El coeficiente de variación total menor que 2 % demostró que el método presenta buena reproducibilidad.
La curva de recuperación mostró un comportamiento lineal debido al valor del coeficiente de correlación cercano a la unidad. La exactitud y linealidad del método se comprobaron mediante las pruebas estadísticas realizadas, ya que el valor de la pendiente no difiere significativamente del valor unitario y el valor del intercepto no difiere significativamente de cero. La recuperación media cumplió con el criterio de aceptación para este tipo de análisis, lo cual coincide con los trabajos publicados por otros autores.5,6 Los resultados permitieron concluir que el método es preciso, exacto y lineal en el intervalo de concentración estudiado.
De la figura 1 se puede concluir que no existe interferencia ni de los excipientes ni de sus productos de degradación, ya que estos eluyen a menores tiempos de retención que el dextrometorfano, por lo que el método resultó ser específico con respecto al placebo y a sus productos de degradación.
El dextrometorfano resultó ser estable en medio ácido; sin embargo, la hidrólisis en medio básico mostró una degradación acentuada, lo cual permite concluir que el principio activo se degrada fácilmente en esta condición. De igual forma se observó la degradación del medicamento en medio oxidante. Por lo tanto, el método cromatográfico resultó ser específico para cuantificar el principio activo después de la degradación de este.
Los ensayos de identificación y valoración cumplieron con los requisitos de calidad establecidos según USP 23 para el medicamento estudiado. Se incluyen además, el cumplimiento de las características organolépticas, contenido por frasco y pH, según los límites establecidos para medicamentos de administración oral o de uso externo.
Summary
The technique described in the USP 23 was applied to carry out the quality control of the dextrometorphan syrup . The reliability of this method was proved by evaluating the accuracy, exactitude, lineality and specificity. The determination of the organoleptic characteristics, pH and content per bottle were also included as own trials of liquid drugs for oral adminstration or for external use.
Subject headings: DEXTROMETORPHAN/pharmacology; QUALITY CONTROL; HIGH PRESSURE LIQUID CHROMATOGRAPHY; DRUGS QUALITY; COUGH/drug therapy; CHEMISTRY, PHARMACEUTICAL.
Referencias bibliográficas
- Calpena AC, Escribano E, Fernández C. Validación de métodos analíticos. Farm Clin 1990;17(9):749-58.
- United States Pharmacopoeial Convention, USP 23. Dextrometorphan Hydrobromide Syrup. 23 ed. Rockville: Mack Printing, 1995:482-3.
- Martindale, The Extra Farmacopoeia. 30 ed. Londres. The Pharmaceutical Press, 1993:746.
- Norma Cubana 26-107-1984. Medicamentos líquidos de administración oral o de uso externo. Especificaciones generales de calidad. La Habana, 1984.
- Lau O, Cheung Y. Simultaneous determination of some actives ingredients in cough-cold syrups by gas-liquid chromatography. Analyst 1990;115(10):1349-53.
- Kountourellis JE, Markopoulos CK. High performance liquid chromatography method for the separation and simultaneous determination of antihistamines, sympathomimetic amines and dextromethorphan in bulk drug material and dosage forms. Anal Lett 1991;23(5):883-91.
Recibido: 13 de noviembre del 2001. Aprobado: 20 de diciembre del 2001.
M.Sc. Alfredo Fernández Serret. Centro de Investigación y Desarrollo de Medicamentos. Ave. 26 No. 1605 entre Boyeros y Puentes Grandes, municipio Plaza de la Revolución, Ciudad de La Habana, CP 10600, Cuba.
1 Master en Ciencias. Licenciado en Química. Investigador Auxiliar.
2 Licenciada en Ciencias Farmacéuticas. Hospital Clinicoquirúrgico "Enrique Cabrera."
3 Licenciado en Ciencias Farmacéuticas. Investigador Agregado.
4 Técnica en Tecnología Farmacéutica.

