










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Farmacia
Print version ISSN 0034-7515
Rev Cubana Farm vol.47 no.1 Ciudad de la Habana Jan.-Mar. 2013
ARTÍCULOS ORIGINALES
A validated RP-HPLC method for the determination of Irinotecan hydrochloride residues for cleaning validation in production area
Método RP- HPLC validado para la determinación de residuos de Irinotecan Hidrocloruro para la validación de la limpieza en el área de producción
P. Sunil ReddyI, K. Sudhakar BabuII, Navneet KumarIII
I Master of Science in Chemistry, Dr. Reddy's Laboratories Ltd., IPDO, Bachupally, Hyderabad-500072, A.P, India.
II Doctor of Philosophy in Chemistry, Department of Chemistry, S. K. University, Anantapur-515055, A.P., India.
III Master of Science in Pharmaceutical Chemistry, Dr. Reddy's Laboratories Ltd., IPDO, Bachupally, Hyderabad-500072, A.P, India.
ABSTRACT
Introduction: cleaning validation is an integral part of current good manufacturing practices in pharmaceutical industry. The main purpose of cleaning validation is to prove the effectiveness and consistency of cleaning in a given pharmaceutical production equipment to prevent cross contamination and adulteration of drug product with other active ingredient.
Objective: a rapid, sensitive and specific reverse phase HPLC method was developed and validated for the quantitative determination of irinotecan hydrochloride in cleaning validation swab samples.
Method: the method was validated using waters symmetry shield RP-18 (250mm x 4.6mm) 5 µm column with isocratic mobile phase containing a mixture of 0.02 M potassium di-hydrogen ortho-phosphate, pH adjusted to 3.5 with ortho-phosphoric acid, methanol and acetonitrile (60:20:20 v/v/v). The flow rate of mobile phase was 1.0 mL/min with column temperature of 25°C and detection wavelength at 220nm. The sample injection volume was 100 µl.
Results: the calibration curve was linear over a concentration range from 0.024 to 0.143 µg/mL with a correlation coefficient of 0.997. The intra-day and inter-day precision expressed as relative standard deviation were below 3.2%. The recoveries obtained from stainless steel, PCGI, epoxy, glass and decron cloth surfaces were more than 85% and there was no interference from the cotton swab. The detection limit (DL) and quantitation limit (QL) were 0.008 and 0.023 µg ml-1, respectively.
Conclusion: the developed method was validated with respect to specificity, linearity, limit of detection and quantification, accuracy, precision and solution stability. The overall procedure can be used as part of a cleaning validation program in pharmaceutical manufacture of irinotecan hydrochloride.
Keywords: cleaning validation, Irinotecan, HPLC-UV, residues, swab analysis.
RESUMEN
Introducción: la validación de la limpieza forma parte integral de las buenas prácticas de manufacturas actuales en la industria farmacéutica. El objetivo principal de la validación de la limpieza consiste en probar la efectividad y la consistencia de la limpieza en un equipo de producción farmacéutica dado para prevenir la contaminación cruzada y la adulteración del producto farmacéutico con otro ingrediente activo.
Objetivo: se desarrollo y validó un método rápido, sensible y específico de fase de reversión HPLC para la determinación cuantitativa del Irinotecan Hidrocloruro en la limpieza de las muestras de frotis de validación.
Método: el método fue validado usando una Columna RP-18 (250mm x 4.6mm) en una columna de 5 µm con una fase móvil isocrática que contenía una mezcla de 0,02 M de potasio dihidrógeno ortofosfato, un PH ajustado a 3.5 con ácido ortofosfórico, metanol y acetonitrilo (60:20:20 v/v/v). La velocidad del flujo de la fase móvil fue de 1.0 mL/min con una columna de temperatura de 25 o C y una detección de longitud de onda de 220 nm. El volumen de inyección de la muestra fue de 100 µl.
Resultados: la curva de calibración fue lineal con una gama de concentración de 0.024 a 0.143 µg/mL y con un coeficiente de correlación de 0.997. La precisión intra y entre días expresada como derivación estándar relativa estuvo por debajo del 3.2 %. Las recuperaciones obtenidas del acero inoxidable, el PCGI, el epoxi el vidrio y de las superficies de tela de Dacrón representaron más del 85 % y no hubo interferencia con los palillos de algodón. Los límites de detección (LD) y de cuantificación (LC) fueron de 0.008 y 0.023 µg ml-1, respectivamente.
Conclusiones: el método desarrollado se validó con relación a la especificidad, linealidad, límite de detección y cuantificación, exactitud y precisión y estabilidad de la solución. El procedimiento general se puede utilizar como parte de un programa de validación de limpieza en la producción farmacéutica del Irinotecan Hidrocloruro.
Palabras clave: validación de la limpieza, irinotecan, RP-HPLC, residuos, análisis por frotis.
INTRODUCTION
Pharmaceutical manufacturing equipment and area has to be cleaned after production in order to avoid cross contamination in the next batch of a different product. The effectiveness of the cleaning process has to be confirmed by cleaning validation, which involves sampling and testing for acceptable residue on the pharmaceutical manufacturing equipment and production área.1
According to FDA guideline, there are two general types of sampling that have been found acceptable: The most desirable direct sampling from the surface of the equipment by using swab and the use of rinse solution.2 Challeneges for cleaning validation are encountered especially when developing an adequate sampling procedure and sensitive analytical methods capable of detecting traces of active pharmaceutical ingredients, which are likely to remain on the surface of the pharmaceutical equipment after cleaning. HPLC coupled with UV detection is widely used to monitor the efficiency of the cleaning methods due to its high sevsititive, selective and automation characterstics.
The aim of this study was to validate simple RP-HPLC method for the quantitative determination of irinotecan hydrochloride residues in production area equipments and to confirm the efficiency of cleaning procedure.
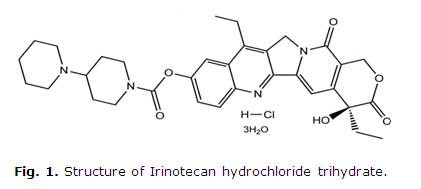
Irinotecan hydrochloride [(S)-4,11-diethyl-3,4,12,14-tetrahydro-4-hydroxy-3,14 -dioxo1Hpyrano[3',4':6,7]-indolizino[1,2-b]quinolin-9-yl-[1,4'bipiperidine]-1'-carboxylate, monohydrochloride, trihydrate] (Fig. 1), a semisynthetic water-soluble derivative of camptothecin, an alkaloid isolated from Camptotheca acuminate,3 has unique antitumor activity, preventing DNA synthesis by inhibiting topoisomerase I.4
Some LC methods have been published for determination of irinotecan in pharmaceutical preparation 5,6,7 and human plasma.8,9 Reported LC methods are not enough sensitive to quantitate trace level residues of irinotecan hydrochloride. A literature survey revealed that no validated cleaning method for irinotecan hydrochloride is to be found. Hence, we have developed a RP-HPLC method for the estimation of trace level residue of irinotecan hydrochloride on swab and rinse solution collected from manufacturing surfaces and production area after cleaning of the equipments. The developed analytical method was validated with respect to specificity, linearity, precision, accuracy, limit of detection (LOD) and quantification (LOQ). These studies were performed in accordance with established ICH guidelines.
METHODS
Chemicals and reagents
The certified irinotecan hydrochloride trihydrate, working standard was supplied by Dr. Reddy's laboratories limited, Hyderabad, India. The HPLC grade acetonitrile and methanol, analytical grade KH2PO4 and ortho-phosphoric acid were purchased from Merck, Mumbai, India. Swabs for sampling were purchased from ITW Texwipe (Philippines).
Equipment
The chromatography analysis was performed using Waters Alliance 2695 separation module (Waters Corporation, Milford, USA) equipped with 2489 UV/visible detector, degasser, quaternary pump and auto sampler system. The output signals were monitored and processed using Empower 2 software. The pH of the solutions was measured by a pH meter (Mettler-Toledo, Switzerland).
Chromatographic Conditions
The method was developed using Waters Symmetry Shield RP-18 (250 mm x 4.6 mm), 5µm particle size column with isocratic mobile phase containing a mixture of 0.02 M potassium di-hydrogen ortho-phosphate, pH adjusted to 3.5 with ortho-phosphoric acid, methanol and acetonitrile (60:20:20 v/v/v). The flow rate of the mobile phase was set at 1.0 mL/min. The column temperature was maintained at 25° C and the eluted compound was monitored at the wavelength of 220 nm. The sample injection volume was 100 µl.
Standard solution preparation
Diluent was prepared by mixing milli-Q water and methanol in the ratio of 20:80 v/v, respectively. An appropriate amount of irinotecan hydrochloride trihydrate was dissolved in diluent to get a stock solution containing 1 mg/mL drug. The final concentration of solution was 0.048 µg/mL of irinotecan hydrochloride trihydrate.
Sample preparation
The selected surfaces (25 cm x 25 cm) of stainless steel, glass, PGCI, epoxy and decron cloth, previously cleaned and dried, were sprayed with 1000 µL of standard solution, for the positive swab control at all concentration level and the solvent was allowed to evaporate. Except decron cloth, all surfaces were wiped with wet cotton swab soaked with extraction solution (water-methanol 20:80, v/v) to remove the residue from the surface. The swabs were placed in the 25 mL screw-cap test tubes containing 10 mL extraction solution. The tubes were placed in an ultrasonic bath for 15 minutes and the solutions were analysed by HPLC.
Rinse-sampling was performed with extraction solvent for decron cloth. The volume of the rinsing liquid for sampling point was 10 mL for 625 cm2 surface.
Validation of proposed method
The method validation was performed in accordance with ICH guidelines.14 The following validation characteristics were addressed: specificity, accuracy, precision, limit of detection and quantification, linearity, range and solution stability.
Specificity
To prove that the determination of active residue is selective and free from anyy disturbing effects, reference solution, blank and spiked solution sampled from stainless stell, glass, PCGI, epoxy and decron cloth surfaces and placebo solution were injected.
Linearity
Linearity of the method was established by analyzing standard solutions at ten different concentration levels i.e. 0.024, 0.036, 0.048, 0.060, 0.072, 0.096, 0.108, 0.119, 0.131 and 0.143 µg/mL.
Limits of Detection (LOD) and Quantification (LOQ)
The LOD and LOQ were determined based on signal-to-noise ratio of 3:1 and 10:1, respectively, by injecting a series of dilute solutions of analyte with known concentrations.
Precision
The precision of the method was evaluated by repeatability and intermediate precision. The repeatability was determined by analyzing six replicated of extraction-recovery samples and expressed in terms of % RSD. The intermediate precision of the method was evaluated using different analyst and different instrument in the same laboratory.
Accuracy
The accuracy of the method was determined in triplicate by spiking all surfaces with known amount irinotecan hydrochloride. The accuracy of the method was checked at three concentration levels, i.e. at 50 %, 100 % and 150 % level. Accuracy is expressed as percentage of standard recovered from sample matrix.
Stability of analytical solutions
The stability of the irinotecan hydrochloride in the swab matrix and standard solution, were tested. The spiked samples and standard solution were stored at bench top and analyzed against freshly prepared standard solution at 24 hrs interval.
RESULTS
Specificity
The figure 2 shows the results of the specificity of the method. As observed in the chromatograms, no sources of interference were observed at the retention time of the analyte.
Linearity
The correlation coefficient was found to be 0.998, demonstrated the excellent relationship between peak area and the concentration of irinotecn hydrochloride.
Limits of Detection (LOD) and Quantification (LOQ)
The LOD and LOQ for irinotecan hydrochloride were found to be 0.008 and 0.023 µg/mL, respectively. At LOQ level, RSD of the irinotecan area from six replicate injections of standard solution was found to be 5.8 %.
Precision
The results of the precision of the method are reported in table 1. In the repeatability and intermediate precision study coefficient of variation was less than 8.1 % and 8.7 %, respectively.
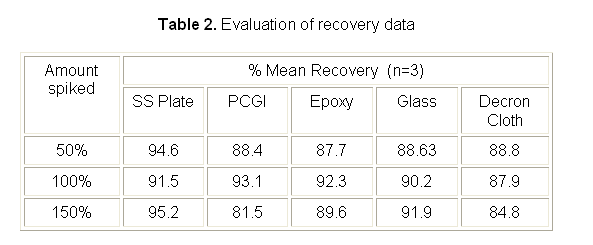
Accuracy
Table 2 shows the results of the accuracy study of the method. The average recovery was between 84.8 to 95.2 % on all surfaces.
Stability of analytical solutions
The variability in the estimation of irinotecan hydrochloride was within + 10 % during solution stability. The results from solution stability experiments confirmed that sample solution and standard solutions were stable up to 48 hrs.
DISCUSSION
Establishing cleaning limits
The acceptable limit for the drug residue must ensure the absence of cross contamination for subsequent batches manufactured in affected equipment [10]. FDA's guidance for determining residue limits requires a logical, practical, achievable and verifiable determination practice.2
The basic principle of cleaning verification/ validation is that the patient should not take more than 0.1 % of the standard therapeutic dose (effective dose). The calculation formula is based on the dosage criteria.11,12
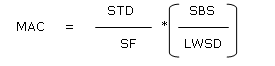
MAC is the maximum allowable carryover, STD is the minimal daily dose (active weight) of previous product, SF is a safety factor (10000), SBS is the smallest batch size of the subsequent product and LWDS is the maximum daily dose (product weight) of the following product.
An additional criterion is the 10 ppm (part per million) limit.13 According to this criterion not more than 10 ppm of the previously manufactured product is allowed to appear in the subsequent product. If the value, which is obtained from the calculation based on the dosage criterion, is greater than 10 ppm, then the 10 ppm criterion is applicable. The acceptable limit for residues (LA) is expressed in µg/dm2.
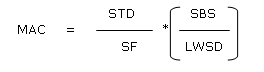
LA is the acceptance limit, A is the sampling area, R is the recovery of the sampling method and TA is the total production line area. On the basis of aforementioned discussion the acceptance limit for the residue of irinotecan hydrochloride is 0.05 ppm.
No sources of interference were observed at the retention time of the irinotecan hydrochloride (Fig. 1), which proved that the method is specific for the quantification of analyte.
To establish linearity, the peak area versus concentration data was treated by linear regression analysis. The correlation coefficient was found > 0.997, demonstrated that the method is linear over the stated range.
The LOD and LOQ for irinotecan hydrochloride were found to be 0.008 and 0.023 µg/mL, respectively. RSD of the area at precision at LOQ level was found to be 5.8 %. Lower values of LOD and LOQ demonstrated that the method is enough sensitive to quantify trace level amount of irinotecan hydrochloride.
The results from precision (table 1) and accuracy (table 2) study confirmed that the method is adequately precise and accurate for the quantification of irinotecan hydrochloride residue on production area equipments.
The sample solution and standard solutions were stable up to 48 hrs. During the stability studies no additional peaks developed and no change in the chromatography of the stored samples and standard were found.
CONCLUSIONS
The proposed method for quantitative determination of irinotecan hydrochloride residue on production area equipments is efficient and sensitive. Validation studies showed that the HPLC-UV method is selective, linear, precise, accurate and robust. The recoveries obtained from stainless steel, PCGI, epoxy, glass and decron cloth surfaces were more than 85 % and there was no interference from the cotton swab. The overall procedure can be used as part of a cleaning validation program in pharmaceutical manufacture of irinotecan hydrochloride.
Acknowledgement
The authors are thankful to the management of Dr. Reddy's Laboratories Ltd., Hyderabad for providing facilities to carry out this work.
REFERENCES
1. PDA Technical Report No. 29, Points to consider for cleaning validation, PDA J. Pharm. Sci. Technol. 52 (1998)1-29.
2. Guide to inspections validation of cleaning processes, U.S. Food and Drug Administration, Office of Regulatory Affairs, Washington, DC, 1993, pp. 1-6.
3. RxList. The Internet Drug Index. Camptosar Injection. [cited 2012 Jan 31]. Available from: http://www.rxlist.com/camptosar_inj-drug.htm
4. Kawato Y, Aonuma M, Hirota Y, Kuga H, Sato K. Intracellular role of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11, Cancer Res., 1991; 51: 4187-4191.
5. Kumar VK, Raju NA, Rani N, Rao JVLNS, Satyanarayana T. The estimation of irinotecan HCl in parenterals by RP-HPLC, Asian J. Research Chem. 2009; 2: 54-56.
6. Balaram VM, Rao JV, Ramakrishna S, Ganesh GS, Krishna TB. Validated reverse phase HPLC method for the determination of Irinotecan in pharmaceutical dosage forms, E-J Chem., 2007; 4: 128-136.
7. Mohammadi A, Esmaeili F, Dinarvand R, Atyabi F, Walker RB. Simultaneous determination of irinotecan hydrochloride and its related compounds by high performance liquid chromatography using ultraviolet detection, Asian J. Chem. 2010; 22: 3966-3972.
8. Doods HM, Robert J, Rivory LP. The detection of photo degradation products of irrinotecn (CPT-11, Campto®, Camptosar®), in clinical studies, using high-performance liquid chromatography/atmospheric pressure chemical ionisation/mass spectroscopy, J. Pharm. Biomed. Anal. 1998; 17: 785-792.
9. Khan S, Ahmed A, Guo W, Wang YF, Abu-Ware A, Ahmad I. A simple and sensitive LC/MS/MS assay for 7-ethyl-10-hydroxycamptothecin (SN-38) in mouse plasma and tissues: application to pharmacokinetic study of liposome entrapped SN-38 (LE0SN38), J. Pharm. Biomed. Anal. 2005; 37: 135-142.
10. Forsyth RJ, Haynes DV, Cleaning validation in pharmaceutical research facility, Pharm. Technol. 1998; 22: 104-112.
11. Guidance on aspects of cleaning validation in active pharmaceutical ingredient plants, Active Pharmaceutical Committee (APIC), 1-56.
12. LeBlance DA, Establishing scientifically justified acceptance criteria for cleaning validation of finished drug products, Pharm. Technol. 1998; 22 (10): 136-148.
13. Fourman GL, Mullen MV, Determining cleaning validation acceptance limits for pharmaceutical manufacturing operations, Pharm. Technol. 1993; 17 (4): 54-60.
14. ICH Q2 (R1), Validation of Analytical Procedures: Text and Methodology, 2005.
Recibido: 16 de agosto de 2012.
Aprobado: 4 de octubre de 2012.
P. Sunil Reddy.
Tel # +91 9963029535; Fax # + 91 40 44346285. E-mail address: sunilpsr@yahoo.com, sunilrp@drreddys.com

{kind=link}
{kind=link}
{kind=link}